リードを表示して塩基置換を見たいときーたとえばSNP解析であったり、A to I editing解析であったり、メチル化解析であったりするときに、とりあえずサルでもできるマッピングをして、IGVでbamファイルを開いたときに出てくるこの画面。
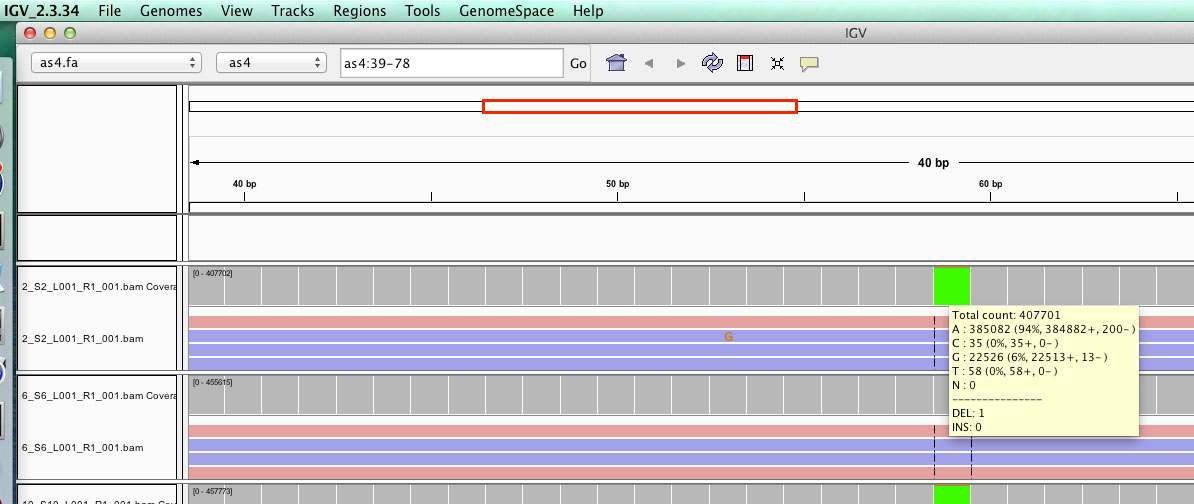
リファレンスゲノムと同じ塩基であれば灰色、設定したthresholdよりも大きい値で他の塩基であればその塩基の色で表示してくれるCoverageのトラックがあるわけですが、そこにカーソルをスクロールオーバーすると、各塩基の組成をポップアップで表示してくれます。ふむふむなるほど、この塩基はA to Gの置換が6%かと、たちどころにわかるのは良いのですが、いざこのデータをexportしたいとなると、途端に壁が立ちはだかります。感覚的にはexportのボタンがどこかにあってそれを押したらオプションが出てきて、、、なんですが、濡れ鼠研究者向けに作られていないIGVにはそのようなボタンはありません。
SNP allele frequency countあたりでググりゃすぐ答えが出るだろうと甘く見積もっていたのですが、samtools mpileupというコマンドでなんとかなりそうなんですが、なんとかなりそうでなんとかならない。ええいこんなんで半日潰すぐらいなら、手で書き写せばいいやろ、、、というのが僕の発想ではあるのですが、たった100塩基でも結構な作業です(本当にやるなよ、、、)。泣きそうになっていたら同じ悩みを持っている研究者はやはりたくさんいるようでこんな掲示板のやりとりが目に入りました。
まあ、よくよく考えてみればIGVで表示されているのだからその機能を使えばすぐにそのデータは取り出せるわけで、、、igvtoolsを例のhomebrewでmacにインストール(リンク記事の下の方)してあれば一発らしいです。
igvtools count -w 1 --bases <input file.bam> <output file.wig> <reference genome.fa>
ここで、-w 1はスキャンするウィンドウで、1塩基ごとであれば1、--basesはよくわからないけど、塩基組成がこれで出てくる、output fileは.wigにしないとバイナリーの記号で出てくるのでわけわからない、という、なんとも濡れ鼠研究者なりの解釈で申しわけありませんが、このコマンドを打って生成されたwigファイルをテキストエディットで開くと、、、
track type=wiggle_0 #Columns: Pos, Combined Strands A, Combined Strands C, Combined Strands G, Combined Strands T, Combined Strands N variableStep chrom=as4 span=1
1 41.0 80.0 145.0 405517.0 0.0
2 35.0 90.0 13.0 407137.0 0.0
3 406171.0 57.0 42.0 1157.0 0.0
4 407187.0 73.0 43.0 199.0 0.0
5 240.0 22.0 406351.0 205.0 0.0
6 205.0 24.0 407154.0 182.0 0.0
........
おお!!これ、これ、まさにこれ、欲しかったのはこのファイル、ということで、無事各塩基のリードカウントがずらりと出てきたファイルが作られていました。あとはexcelにコピペして適当にグラフを作るだけ。 IGV上で見た目おおっと思ったことは、とりあえずIGVのGUIか、igvtoolsでできるらしい(もっと高度なこともできるらしい)ということでした。
しかしそれにしてもネット上、あるいはWiki関連の、NGS関連technical tip(というほどのものではないのかもしれませんが)情報は実に充実しています。先輩の手つきを背中越しに見てその技をぬすめ!!という文化からすると隔世の感がありまする。
ーー
以下、前記事のアップデート情報こちらにも載せておきます。
リードをそのままマッピングしてもそこそこの結果が出てきますが、特に、塩基置換を調べる時など、クオリティーが低いリードを無理やりマッピングしてしまうとノイズが増えてしまうようです。クオリティーフィルターはいろいろありますが、FASTX-Toolkitが便利ということでインストールしようとしても動かず、Macでは動かないのかなということで敬遠していたのですが、最近チェックしてみると、今ではhomebrewで普通にインストールできるようです。というわけで、たとえば3' 末端からクオリティが 30 未満の塩基をトリミングし、長さが 100 bp 未満となったリードを破棄する。リードの 80% 以上の塩基がクオリティ 20 未満のリードならば除去する。というフィルターならば、
fastq_quality_trimmer -t 30 -l 100 -i <inputのファイル.fastq> | fastq_quality_filter -q 30 -p 80 -o <outputのファイル.fastq>
でオーケー。FastQCとかでフィルターがうまくいっているかチェックした後マッピングしなおすと、劇的に結果が変わるときもあるようです。

 2020
2020