例えばの話、qPCRのデータを解析する感覚でカウント数を総リード数で割った値のデーターテーブルを作って、平均と標準偏差を出したりt-testを行ったりすれば発現変動遺伝子が同定できそうですが、おっとどっこい。これがなかなか難しいみたいなんですね。マニュアルに詳しく書かれていますが、一言で言ってしまうと、めっちゃ発現量が多い少数の遺伝子の挙動にひきずられて、発現量が低い大多数の遺伝子の発現量が見かけ上変化したように見えてしまうのを補正しなくてはいけない。つまり、シークエンスというのは全体のほんの一部しか読んでいないので、カウント数はあくまでも相対的なrepresentationを表しているだけで、その遺伝子が細胞でどれだけ発現しているかを表す数値にはなっていません。たとえば、10人で飲みに行った時に一人でずーと喋っているAさんがいたとして、Aさんが帰ったからといって残ったBさんが(AさんとBさんの間に直接間接のつながりがなければ)とたんにおしゃべりになる、ということはないと思われますが、Bさんの発言が全体の何割を占めていたか、ということをカウントすると、Aさん退席前後で劇的におしゃべりになったかのように見えてしまう、ということですね。
ともあれ、DEseq2を使ってnormalizationを行ってみます。これもRから使えるのでRStudioに入ります。DESeq2のインストールは
source("https://bioconductor.org/biocLite.R")
biocLite("DESeq2")
library(DEseq2)
だけでおしまい。素晴らしい。次回からはlibrary(DEseq2)だけで使えます。前回作ったカウントデータがはいったリストfcをそのまま引き継いで使います。
# normalize with DESeq2
library(DESeq2)
dt <- as.matrix(fc$counts)
dt1 <- dt[apply(dt,1,sum)>6,]
group <- data.frame(con = factor(c("normal","normal","normal","HFD","HFD","HFD")))
dds <- DESeqDataSetFromMatrix(countData = dt1, colData = group, design = ~ con)
dds <- DESeq(dds)
res <- results(dds)
res$gene_id <- row.names(res)
write.table(res,file="DESeq2_result.txt",sep="\t",row.names=F,col.names=T,quote=F)
やっていることはマニュアル通りで、fcリストの中のcountsデータだけを取り出してmatrix形式のデータdtに格納し、6サンプルの合計のリードが6に満たないような超低発現量の遺伝子は除外し、factor(c("normal","normal","normal","HFD","HFD","HFD")の部分に実験の区分を入力し、あとはDESeqちゃんに頑張ってもらうだけ。発現量の変動の結果はresというデータフレームに入れています。最後に、後々ggplot2などで使うためにid情報をgene_idという列を作って入れ、DESeq2_result.txtというテキストファイルに出力しています。
head(res)でちょっと頭のところを見て見ると、
log2 fold change (MAP): con normal vs HFD
Wald test p-value: con normal vs HFD
DataFrame with 6 rows and 7 columns
baseMean log2FoldChange lfcSE stat pvalue padj gene_id
ENSMUSG00000051951.5 15.012639 0.3847144 0.5359910 0.7177628 0.4729036 0.9165602 ENSMUSG00000051951.5
ENSMUSG00000103377.1 2.154011 -0.9483600 0.6889626 -1.3765043 0.1686656 NA ENSMUSG00000103377.1
ENSMUSG00000103201.1 2.557988 -0.1330612 0.6776607 -0.1963537 0.8443333 NA ENSMUSG00000103201.1
ENSMUSG00000103147.1 4.049702 0.3012301 0.6666157 0.4518797 0.6513556 0.9610050 ENSMUSG00000103147.1
ENSMUSG00000103161.1 1.643521 0.1851621 0.6896544 0.2684854 0.7883257 NA ENSMUSG00000103161.1
ENSMUSG00000102331.1 1.766659 0.3572244 0.6907083 0.5171856 0.6050266 NA ENSMUSG00000102331.1
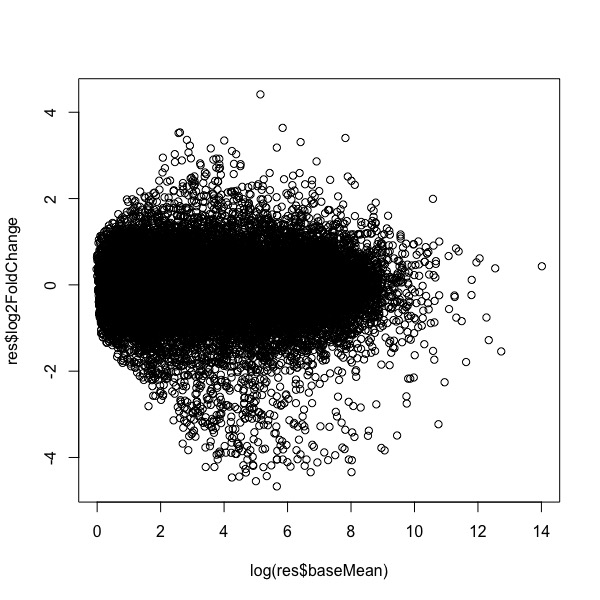
うんうん。いい感じ。baseMeanはnormalizeするために使った発現量の目安、log2FoldChangeとpadjが一番見たいところでそれぞれ発現量の変化と有意差になります。Rstudio組み込みのplot関数を使ってMA-plotやvolcano plotを見てみればだいたいの雰囲気が掴めます。
#MA-plot
plot(log(res$baseMean),res$log2FoldChange)
#volcano plot
plot(res$log2FoldChange,-log(res$padj))

しかしこのリスト、遺伝子名がgene idなので、どれがどれだかさっぱり。やっぱり見慣れたgene symbolの情報が欲しいです。というわけで、最初に使ったgtfファイルから必要なアノテーションを取ってきて、DESeq2で作った発現量の変化のファイルとマージしてやることにします。
gtfファイルをRで使えるデーターテーブルに取り込むには、rtracklayerというライブラリを使うのが便利です(追記:以前この項で"Biostrings"と書いてましたがreadGFFはrtracklayerのコマンドでした。すいません。)。インストールは、
source("https://bioconductor.org/biocLite.R")
biocLite("rtracklayer")
library(rtracklayer)
でオーケー。次回からはlibrary(rtracklayer)だけで(略)
これでreadGFFというコマンドがつかえるようになるので、gtfというデータフレームにカウントの時に使ったgtfデータを取り込み、遺伝子ごとにカウントしたのでgeneの情報だけ抜き出したデータフレームgtf_geneを作ります。
gtf <- readGFF("./genome/gencode.vM17.chr_patch_hapl_scaff.annotation.gtf")
gtf_gene <- subset(gtf, gtf$type == "gene")
gtfにどんな情報が入っているのか最初の数行を見て見ると、、
colnames(gtf_gene)
[1] "seqid" "source" "type" "start"
[5] "end" "score" "strand" "phase"
[9] "gene_id" "gene_type" "gene_name" "level"
[13] "havana_gene" "transcript_id" "transcript_type" "transcript_name"
[17] "transcript_support_level" "tag" "havana_transcript" "exon_number"
[21] "exon_id" "protein_id" "ccdsid" "ont"
なので、gene_idで紐付けしてカウントデータとmergeしてやります。無駄な情報も結構あるので、ここでは遺伝子の名前のgene_name、どんな種類のカテゴリーか(mRNAかlincRNAかとか)の情報が入っているgene_typeだけ取り出しましょうか。resはDEseq2のデータフレームなので通常のデータフレームに変換して、mergeコマンドで紐付け。今回は同じ名前のカラムで紐付けするので、とくに列指定しなくてもうまくいきます。マージした結果はDEseq2_result_ref.txtというファイルで出力しておきます。
gtf_gene_1 <- gtf_gene[,c("gene_id","gene_name","gene_type")]
DEseq2_result <- data.frame(res)
DESeq2_result_ref <- merge(DEseq2_result,gtf_gene_1)
write.table(res,file="DESeq2_result_ref.txt",sep="\t",row.names=F,col.names=T,quote=F)
これで一応目的のファイルができました!あとは適当に遊ぶだけ。log2FoldChangeが3以上のリストを見たければ
DESeq2_result_ref[DESeq2_result_ref$log2FoldChange>3,]gene_id baseMean log2FoldChange lfcSE stat pvalue padj gene_name
39 ENSMUSG00000000253.13 2498.90834 3.403819 0.5064163 6.721386 1.800045e-11 1.741661e-08 Gmpr
1457 ENSMUSG00000015437.5 70.03353 3.102009 0.5814216 5.335214 9.543187e-08 1.621176e-05 Gzmb
2008 ENSMUSG00000019945.10 11.55095 3.029959 0.6711592 4.514515 6.346171e-06 5.061924e-04 RP23-2A21.1
2219 ENSMUSG00000020331.9 287.92907 3.180661 0.5797693 5.486081 4.109493e-08 7.816466e-06 Hcn2
2753 ENSMUSG00000021313.16 171.48167 4.413720 0.5021746 8.789214 1.506097e-18 3.351668e-14 Ryr2
4888 ENSMUSG00000026358.13 345.57631 3.638248 0.5571884 6.529655 6.592116e-11 3.860551e-08 Rgs1
.... .....................と入れれば良いだけだし、Malat1がどうなっているか気になるのであれば
DESeq2_result_ref[DESeq2_result_ref$gene_name == "Malat1",]gene_id baseMean log2FoldChange lfcSE stat pvalue padj gene_name gene_type
19863 ENSMUSG00000092341.2 133735.4 0.1151078 0.4079032 0.282194 0.7777948 0.9795941 Malat1 lincRNAでオッケ。動作が非常にキビキビしているので慣れるとexelよりははるかに楽チンですね。RStudioでやれば何をどのように見ていったかの思考過程も全てログに残せるので、とても便利。仕事の記録はすべて残しておくに越したことはありません。
はてさて。先ほどのプロットの図ですが、なんとも味気ないので、少し綺麗にggplot2で書いて見ます。また、せっかくメタ情報も取ってきているので、protein_codingの遺伝子とlinc_RNAの遺伝子を色付けしてみました。これが結構面白い。おーっ!色がついた!とか、lincRNAでもmRNAでもないこの点はなんなんだとか。アルファをかけて半透明にするとなんか賢くなった気がするのが不思議。ggplot2で凝りだすとあっという間に時間が過ぎてしまい会議があるのも忘れて没頭してしまったりするわけですが、はい、すいません。次回から気をつけます。。。
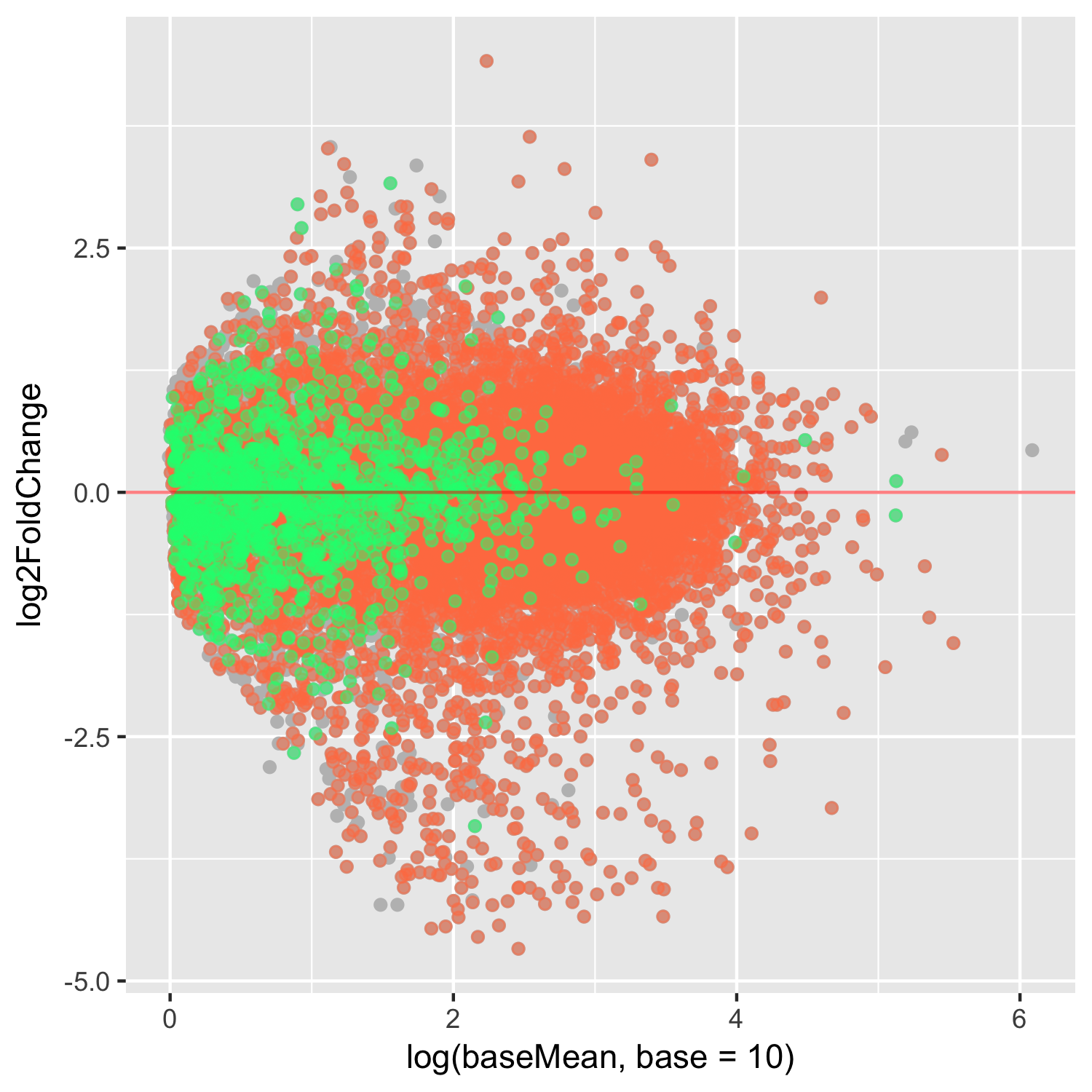
ここでのggplotのポイントを簡単に。散布図などはgeom_pointで書けます。どのデータフレームを使うかは、data = で指定。x軸、Y軸にどのカラムを使うかはaesで指定。あらかじめ色々な条件(メタ情報、発現量、p値etc)で絞り込んだデータフレームを作って異なる色で重ね合わせができます。色指定はcolor=。alpha=で半透明にできます。プロットが重なる時に便利。色見本は奥村研究室のサイトに詳しいです。この場合、上の4行で描画はおしまい。あとのmodify plotはパネルのお化粧。共通にしておくと論文間で統一感のある図が作れます。ggsaveで保存。コンソールにgと打ち込めば今どんな図が書けてるかplot画面でチェックできます。
#ggplot2
library(ggplot2)
#select subset of genes
all <- DESeq2_result_ref
mRNA <- DESeq2_result_ref[DESeq2_result_ref$gene_type == "protein_coding",]
lincRNA <- DESeq2_result_ref[DESeq2_result_ref$gene_type == "lincRNA",]
#drow MA plot
g <- ggplot(NULL)
g <- g + geom_point (data = all, aes (x = log(baseMean,base = 10), y = log2FoldChange), color = "gray")
g <- g + geom_point (data = mRNA, aes (x = log(baseMean,base = 10), y = log2FoldChange), color = "coral", alpha = 0.5)
g <- g + geom_point (data = lincRNA, aes (x = log(baseMean,base = 10), y = log2FoldChange), color = "springgreen", alpha = 0.5)
#modify plot
g <- g + geom_hline(yintercept = 0,colour="red",alpha = 0.5)
g <- g + theme(axis.text.x = element_text(size = 18),axis.title.x=element_text(size=20)) + #X label size
theme(axis.text.y = element_text(size = 18),axis.title.y=element_text(size=20)) + #Y label size
theme(plot.title = element_text(size=22)) + #title size
ylab("log2 (HFD/control) ") + # X label
xlab("log10 (baseMean)") +# Y label
labs(title = "gene expression changes upon HFD feeding") #title
ggsave(filename = "MA_plot_Neuro2a_genes.png",g,width = 5, height = 5)

 2020
2020