まずは、リボゾーム遺伝子のbedファイルをとってきます。いろんなやり方があると思いますが、UCSC Browserのtable browserhttps://genome.ucsc.edu/cgi-bin/hgTablesから取ってくるのが一番手っ取り早いですね。
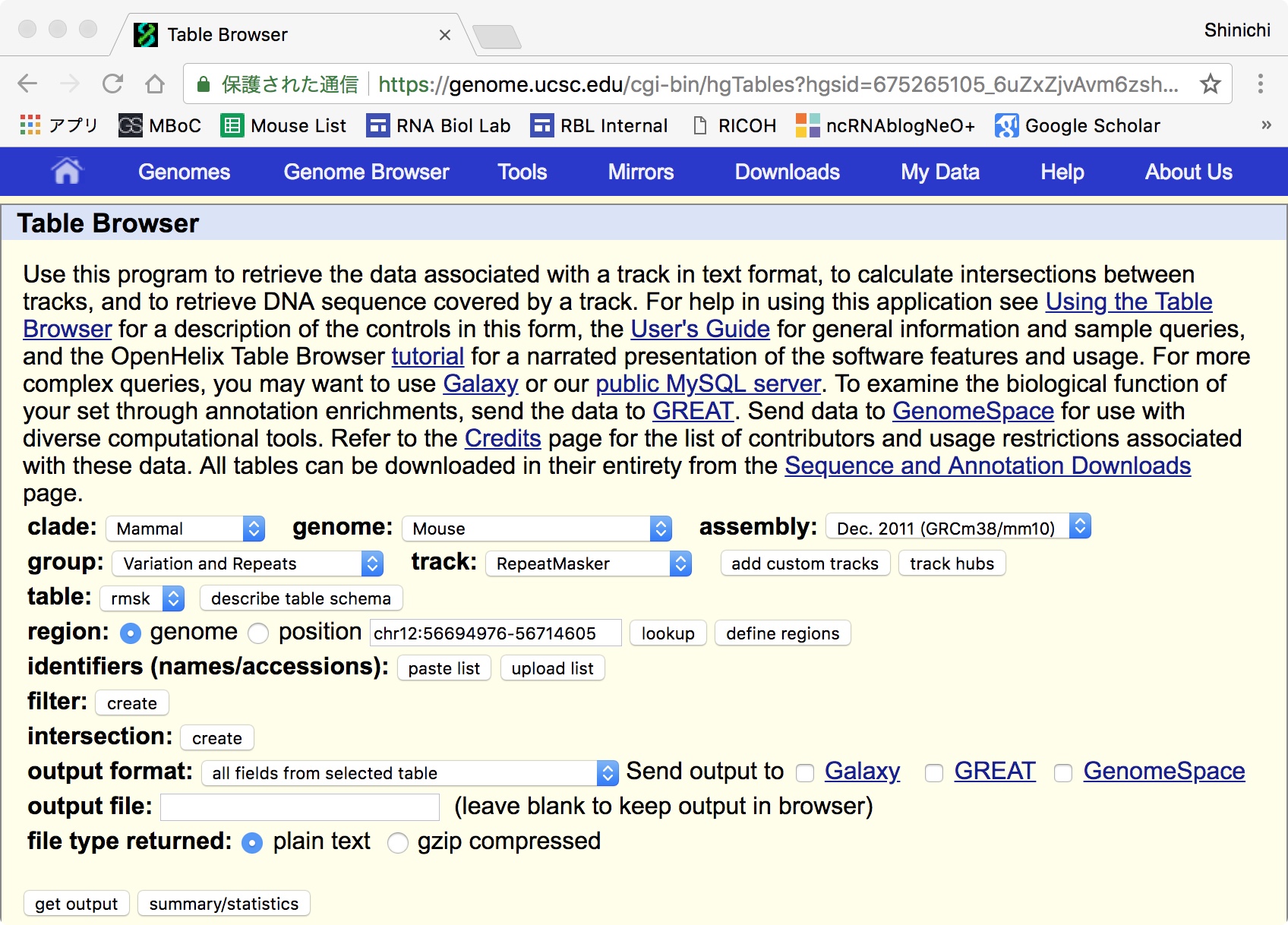
プルダウンメニューで
group: Variation and Repeats
track: RepeatMasker
table:rmsk
と設定して、filterのcreateボタンを押します。
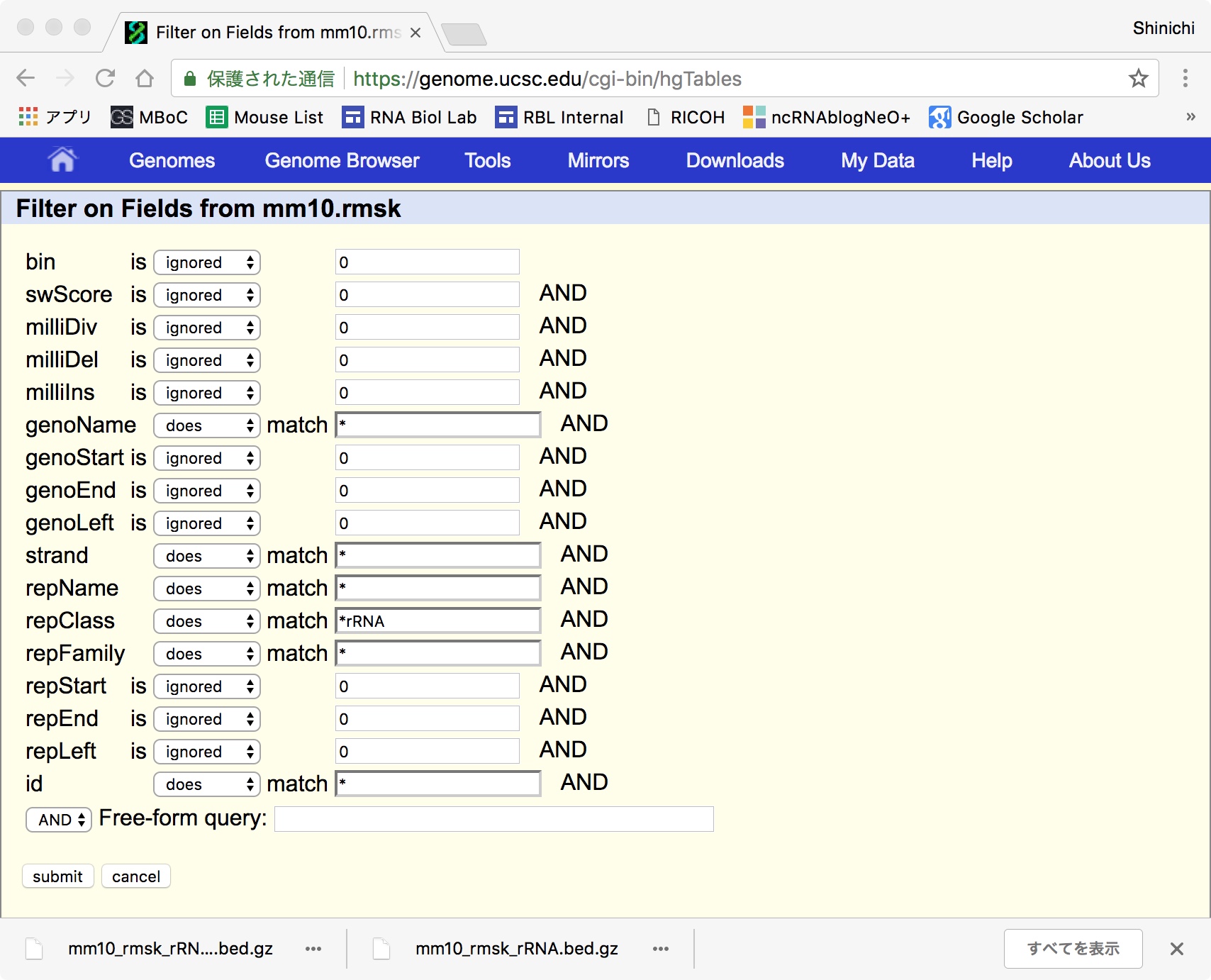
するとこんな画面になるので、repClassのところにrRNAをいれてsubmitボタンを押して先ほどの画面に戻ります。あとはoutput fileのところに自分の好きな名前(ここではmm10_rmsk_rRNA.bed)を入れてget outputボタンを押せばダウンロードできます。大した大きさのファイルではないので下の方のリンクにも入れておきました。このファイルを前回使ったgenomeフォルダの中に入れておきます。
あとはbedtoolsのintersectコマンドを使ってこのbedファイルにかぶってくるリードを取り除くだけ。取り除いたファイルは新しいbamファイルSRR6946223.sort_rRNA_rm.bamとして出力させてます。
cd /Users/Nakagawa_imac/Desktop/2018_NGS_standard
intersectBed -abam SRR6946223.sort.bam -b ./genome/mm10_rmsk_rRNA.bed -v > SRR6946223.sort_rRNA_rm.bam
さて、次はこのファイルをリード数をカウントしてくれるツールに投げるわけですが、手っ取り早く遺伝子ごとの発現量の差を見るだけであったらRsubreadパッケージの中のfeatureCountsがお手軽です。ここから先は基本Rのコンソールでの操作になります。インストールはRStudioのセッションを立ち上げて、
source("https://bioconductor.org/biocLite.R")
biocLite(pkgs="Rsubread")
library ("Rsubread")
で完了。2回目からはlibrary("Rsubread")だけで呼び出せます。遺伝子のアノテーション情報はfeatureCountsに組み込まれているものがあるのですが、やはりこれも最新のが良いでしょうということで、GENCODEのページから取ってきます。マウスだとgencode.vM17.chr_patch_hapl_scaff.annotation.gtfが最新ですね。これもダウンロードして、作業フォルダの中のgenomeフォルダの中に入れておきます。
あとは基本的にはデフォルトの設定で走らせれば良いのですが、一点だけ留意した方が良いのがO (allowMultiOverlap)オプションです。これは、アノテーションが重なっている領域にあるリードを数えるかどうか、ということなのですが、デフォルトでは遺伝子ごとの発現量を特異的にカウントするためにオフになっています。たとえば下図のcase1のようなときは遺伝子A、Bにそれぞれ特異的な領域があるので緑の色がかけられていない領域にマップされたリードをカウントすることでサンプル間の遺伝子A、Bの発現量の変化を正確に比較することができますが、case2のような時は、遺伝子Aの発現量がゼロになってしまうんですね。
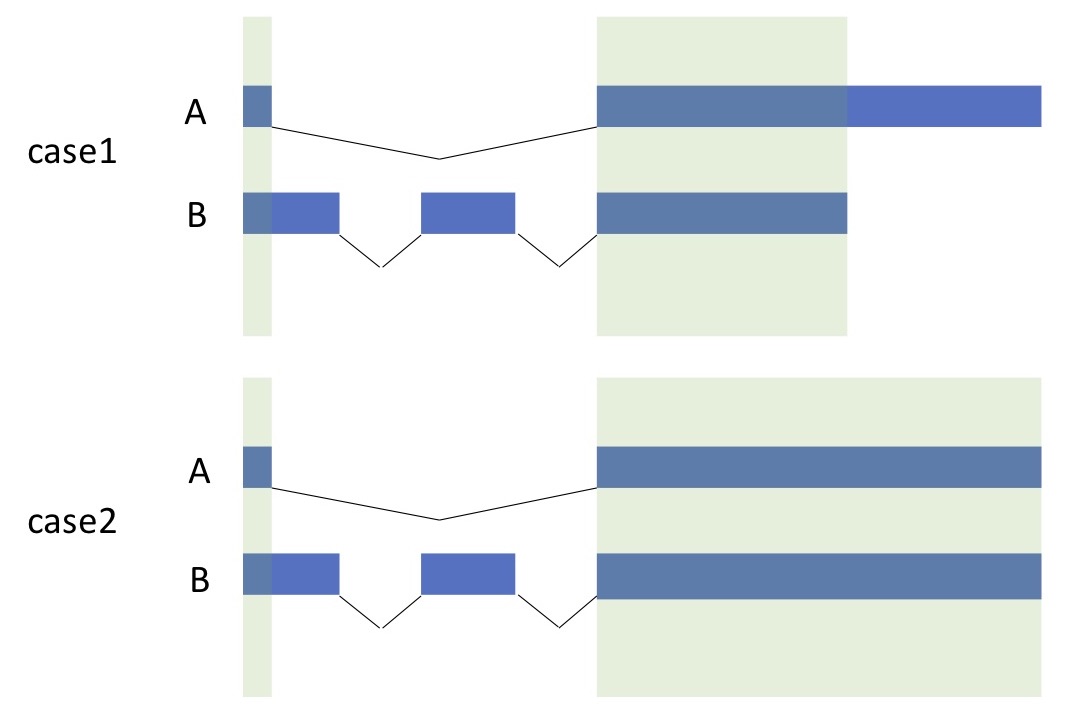
Neat1_1とNeat1_2の場合がまさにそうで、デフォルトだとNeat1_1の発現はゼロ。そんなわけはないでしょう、ということで、このオプションをオンにすると、重なった領域に張り付いたリードを等分してくれます。もちろん、本来は発現していないのに発現していると勘違いされることもあるわけで、そこは個別に留意が必要。入力するbamファイルがSRR6946223.sort_rRNA_rm.bamから連番で6つのファイル、遺伝子のアノテーションファイルがgencode.vM17.chr_patch_hapl_scaff.annotation.gtf、iMacの4スレッドをフルに使うとすると、これまたRStudioで
setwd("~/Desktop/2018_NGS_standard")
fc <- featureCounts (files = c("SRR6946223.sort_rRNA_rm.bam",
"SRR6946224.sort_rRNA_rm.bam",
"SRR6946225.sort_rRNA_rm.bam",
"SRR6946226.sort_rRNA_rm.bam",
"SRR6946227.sort_rRNA_rm.bam",
"SRR6946228.sort_rRNA_rm.bam"),
annot.ext="./genome/gencode.vM17.chr_patch_hapl_scaff.annotation.gtf",
isGTFAnnotationFile=TRUE,
nthreads=4,
allowMultiOverlap = TRUE)
write.table(file = "counts.txt", fc$counts, quote=FALSE, sep="\t")
とすればオーケー。この場合、全てのカウントデータを格納したfcというリストが作られます。これはそのまま次のDEseq2で使用できるので便利。一応個々の実験のカウントデータが欲しい時も出てくるかと思うので、最後のwrite tableでタブ区切りテキストとして保存しています。
featureCountsも爆速で、single endであれば1分ぐらいでカウント終了!Cufflinksを使って数時間かかっていたことを考えるとありえない速さ。もちろんtranscriptごとのカウントができていないという大きな欠点はありますが、ちょっと発現見てみよう、というときは十分です。過ぎたるは及ばざるがごとし。コバエの退治にロケット砲をつかう必要はありません。ただ、pair-endになるとちょっと事情がかわってきて、なぜかbamファイルを一旦samファイルに変換しているみたいで、10分ぐらいはかかってしまいます。発現量のカウントだけならpair-endのsraファイルを最初からsingle endのリードとしてfastqに変換して出てきたファイルをcatでつないでHISAT2に投げたらこの辺り楽なのかしらん。いずれにせよ、Cufflinksに比べればすごく早いので、たとえpair endのデータであってもソフトボール大会に備えて外でキャッチボールでもしてくれば6サンプルぐらいすぐにカウントしてくれます。
お次はカウントデータの標準化と可視化です。

 2020
2020