前回は、Table Browserから転写産物のRNAの長さ、5'UTRの長さ、3'UTRの長さをとってきたファイルを作りました。ところが、このファイル、UCSC gene IDはあるものの、遺伝子名の情報が付いていません。例えば、Cuffidiffが吐き出してきた遺伝子発現変化のファイルgene_expression.diffのどこを探しても、愛しのGomafuちゃんがどこにあるのかさっぱりわかりません。そこで、UCSCのIDとGene Symbolを紐付けしたファイルをTable Browserから落としてきて、これを使って、転写産物の長さの情報、遺伝子発現の変化の情報、各遺伝子名の情報を統合してやります。
まず、遺伝子のアノテーション情報はtable browserの group: Genes and Gene Predictions track: UCSC genes table: kgXref に入っていますのでこれをhg38_info.txtというファイルで落としてきます。今回は見やすくするためにoutput format: selcted fields...で、kgIDとgeneSymbolとrefseqだけ選んでいます。RでkgXrefという名前でこいつを開いてやって、headで内容を確認してやると、、、
> kgXref<-read.delim("hg38_kg_xref.txt")
> head(kgXref)
X.kgID geneSymbol refseq
1 uc031tla.1 MIR6859-1 NR_106918
2 uc057aty.1 RP11-34P13.3
3 uc057atz.1 RP11-34P13.3
4 uc031tlb.1 MIR1302-2 NR_036051
5 uc001aak.4 FAM138A NR_026822
6 uc057aua.1 FAM138A
X.kgIDというラベルの列にUCSCのIDが、geneSymbolの列に遺伝子名が、refseqの列にrefseqのIDがついているのがわかります。まあ、言ってみれは最近流行りのアレですアレ。IDがペンで、geneSymbolとrefseqがリンゴ。

次に、前回作ったRNAの長さなどの情報が入ったhg38_info.txtの内容をinfoに読み込み、中身を確認。
> info<-read.delim("hg38_info.txt")
> head(info)
ID exon length utr5 utr3
1 uc031tla.1 1 68 NA NA
2 uc057aty.1 3 712 NA NA
3 uc057atz.1 2 535 NA NA
4 uc031tlb.1 1 138 NA NA
5 uc001aak.4 3 1187 NA NA
6 uc057aua.1 2 590 NA NA
こちらはIDというラベルの列にUCSCのIDが入っていて、その他の列に転写産物の情報ですね。はい。そうです。IDがペンで、その他の情報がパイナップル。
.jpg)
ここまできたら、もうやること決まってますね。PPAP!
データの紐付けにはmergeという魔法のようなコマンドが用意されています。mergeの後につなぎたいデーターフレームをカンマで指定。次に、それぞれのデーターフレームのどの列を使うのかを、by.x="XXXX", by.y="YYYY"で指定するだけ。all=Tは、mergeした行に欠損値があった場合、その行を含めるか含めないかのオプションで、TはTRUE、すなわち、全て残しておこうね、というオプションになります。by.xやby.yは、列番号で指定することも可能で、この場合であればどちらも1列目なので、by.x=1, by.y=1でも指定可能。
> m1<-merge(kgXref,info,by.x="X.kgID",by.y="ID",all=T)
> head(m1)
X.kgID geneSymbol refseq exon length utr5 utr3
1 uc001aak.4 FAM138A NR_026822 3 1187 NA NA
2 uc001aal.1 OR4F5 NM_001005484 1 918 NA NA
3 uc001aaq.3 RP4-669L17.10 4 696 NA NA
4 uc001aar.3 RP4-669L17.10 3 457 NA NA
5 uc001abp.3 LINC01128 6 874 NA NA
6 uc001abt.5 FAM41C NR_027055 3 1807 NA NA

はい。見事につながりました。
Exelでも同じことは列のコピペでできるわけですが、その前に、紐付けする列をソートしておかないと、紐付けすることができません。mergeのコマンドの優れている点は、いちいちソートしなくても指定した列の値でPPAPできるところですね。
さて、次に、これらの情報をcuffdiffが吐き出してきたgene_expression.diffファイルと統合してみます。まずはgene_expression.diffファイルを読み込んで、列の数が多いので、紐付けするために必要なtest_id及び、その値が意味があるかどうかのstatus, 各条件でのFPKM値であるvalue_1, value_2,発現量の比である log2.fold_change. だけ抜き出してみます。Rだと、<-XX[行,列]で抜き出しができて、行、列、のところはベクトルで複数の値を指定することもできるんですね。全ての行or列を抜き出したい時は、空欄にしておけばオーケー。
> diff<-read.delim("gene_expression.diff")
#抽出する列をヘッダーの名前で指定
> diff1<-diff[,c("test_id","status", "value_1", "value_2", "log2.fold_change.")]
#もしくは列を数字(1列目ならびに7~10列目)で指定
> diff1<-diff[,c(1,7:10)]
> head(diff1)
test_id status value_1 value_2 log2.fold_change.
1 uc001aak.4 NOTEST 0.000000 0.00000 0.00000
2 uc001aal.1 NOTEST 0.000000 0.00000 0.00000
3 uc001aaq.3 NOTEST 0.000000 0.00000 0.00000
4 uc001aar.3 NOTEST 0.000000 0.00000 0.00000
5 uc001abp.3 OK 0.631301 1.46371 1.21323
6 uc001abt.5 NOTEST 0.093125 0.00000 -Inf
ここで、ちょっと問題があります。それぞれのデータフレームの行数をnrow関数で確認してみると、、、
> nrow(m1) [1] 197782 > nrow(diff1) [1] 197597
ん?cuffdiffのファイルでは行数が減っている。。。つまり、幾つかデータが飛んでいるわけですね。プログラムのエラーか仕様なのか、そこはmoistureなのでそっとしておいて、とりあえず対応のつくデータだけ対応させたい。Excelだとvlookup関数など使ってけっこうややこしいのですが、Rなら簡単!mergeが勝手に紐付けできるデータを見つけてきてPPAPしてくれます。
> m2<-merge(m1,diff1,by.x=1,by.y=1,all=t)
> head(m2)
X.kgID geneSymbol refseq exon length utr5 utr3 status value_1 value_2 log2.fold_change.
1 uc001aak.4 FAM138A NR_026822 3 1187 NA NA NOTEST 0.000000 0.00000 0.00000
2 uc001aal.1 OR4F5 NM_001005484 1 918 NA NA NOTEST 0.000000 0.00000 0.00000
3 uc001aaq.3 RP4-669L17.10 4 696 NA NA NOTEST 0.000000 0.00000 0.00000
4 uc001aar.3 RP4-669L17.10 3 457 NA NA NOTEST 0.000000 0.00000 0.00000
5 uc001abp.3 LINC01128 6 874 NA NA OK 0.631301 1.46371 1.21323
6 uc001abt.5 FAM41C NR_027055 3 1807 NA NA NOTEST 0.093125 0.00000 -Inf
はい。これでオーケーですね。論文のsupplementのデータなどでは遺伝子名をrefseqのIDでだしているものもあればGeneSymbol、 Ensemble ID、アレイIDでだしているものもあります。直接比較することはできなかったりしますが、Rを使ってmergeで一発。こんな便利なツール、46年間も知らずにいたのが恥ずかしい。
特定の条件のものを取ってくる場合にはsubsetという関数が用意されています。例えば、cuffdiffのgene_expression.diffにstatusという列がありますが、あまりにもリード数が多すぎてカウントできなかった場合はHIDATA、あまりにも発現量が低くてカウントできなかっりアラインメントできなかった場合はLOWDATAやNOTESTもしくはFAILという文字が入れられています。解析の対象となるのはここがOKとなっているものだけですので、それを抽出します。subset(データセット,条件式)でその行だけ抽出することができます。
> m2_filtered<-subset(m2,m2$status=="OK")
> head(m2_filtered)
X.kgID geneSymbol refseq exon length utr5 utr3 status value_1 value_2 log2.fold_change.
5 uc001abp.3 LINC01128 6 874 NA NA OK 0.631301 1.463710 1.213230
9 uc001abz.5 NOC2L NM_015658 19 2790 50 490 OK 85.255800 133.532000 0.647319
10 uc001aca.3 KLHL17 NM_198317 12 2560 107 524 OK 2.969250 3.110510 0.067049
11 uc001acb.2 KLHL17 2 955 NA NA OK 2.436020 0.841683 -1.533180
17 uc001aci.3 HES4 NM_021170 4 899 138 95 OK 53.091600 184.018000 1.793290
18 uc001acj.5 ISG15 NM_005101 2 711 151 62 OK 2.893020 3.766230 0.380546
条件一致は=でなくて==なんですねえ。これで丸一時間ハマッていたことはヒ・ミ・ツ。TBSにHClを入れ忘れてたみたいなもんでしょうか。共通試薬でこれやると罪深いですが、Rなら被害は自分だけ。ちなみに、これは<-XX[行、列]の文を使っても同じことができるみたいです。行の所に、statusがOKという条件式を入れるだけ。
> m2_filtered<-m2[m2$status=="OK",]
> head(m2_filtered)
X.kgID geneSymbol refseq exon length utr5 utr3 status value_1 value_2 log2.fold_change.
5 uc001abp.3 LINC01128 6 874 NA NA OK 0.631301 1.463710 1.213230
9 uc001abz.5 NOC2L NM_015658 19 2790 50 490 OK 85.255800 133.532000 0.647319
10 uc001aca.3 KLHL17 NM_198317 12 2560 107 524 OK 2.969250 3.110510 0.067049
11 uc001acb.2 KLHL17 2 955 NA NA OK 2.436020 0.841683 -1.533180
17 uc001aci.3 HES4 NM_021170 4 899 138 95 OK 53.091600 184.018000 1.793290
18 uc001acj.5 ISG15 NM_005101 2 711 151 62 OK 2.893020 3.766230 0.380546
なんだ。こっちのほうが簡単ではないか。Would you mind if I ask you something...の代わりにFuXX!ですませるようなもんでしょうか。遠回しに言わないで、言いたいことがあったらはっきり言いましょうはっきりと。
データの紐付けができましたから、遺伝子名で発現量の検索をすることもすぐにできます。Gomafuちゃんはどうだったのか知りたかったら、
> m2_filtered[m2_filtered$geneSymbol=="MIAT",] [1] X.kgID geneSymbol refseq exon length utr5 utr3 status [9] value_1 value_2 log2.fold_change. <0 rows=""> (or 0-length row.names)
な、何だ。。。この解析で使った細胞では発現していないんですね。NEAT1ならどうだろう。
> m2_filtered[m2_filtered$geneSymbol=="NEAT1",]
X.kgID geneSymbol refseq length exonCount utr5 utr3 status
37408 uc010rog.3 NEAT1 NR_131012 22743 1 NA NA OK
72300 uc058djn.1 NEAT1 1811 2 NA NA OK
72306 uc058djt.1 NEAT1 483 2 NA NA OK
value_1 value_2 log2.fold_change.
37408 22.6087 2.92937 -2.94822
72300 12.9806 2.45525 -2.40242
72306 36.4155 11.91400 -1.61189
ちゃんと出てきました。
Exelの便利な機能の一つはsortや条件が一致する行の抽出ですが、それもRだとサクサク簡単にいきます。例えば、ノンコーディングRNAだけ抽出したい場合。ちょっと荒っぽいですが、refseqIDがNR_で始まるものをconservativeなノンコーディングRNAの定義として考えることができます。refseqの列にNR_が含まれている行の番号のベクトルを取ってくるgrepというとっても便利な関数があるので、先程の=[行,列]の構文と組み合わせてやるだけ。タンパク質をコードする転写産物はNM_で始まるので、それで取ってくれば良いですね。
> coding<-m2_filtered[grep("NM_",m2_filtered$refseq),]
> head(coding)
X.kgID geneSymbol refseq exon length utr5 utr3 status value_1 value_2 log2.fold_change.
9 uc001abz.5 NOC2L NM_015658 19 2790 50 490 OK 85.25580 133.53200 0.647319
10 uc001aca.3 KLHL17 NM_198317 12 2560 107 524 OK 2.96925 3.11051 0.067049
17 uc001aci.3 HES4 NM_021170 4 899 138 95 OK 53.09160 184.01800 1.793290
18 uc001acj.5 ISG15 NM_005101 2 711 151 62 OK 2.89302 3.76623 0.380546
19 uc001ack.3 AGRN NM_198576 36 7323 50 1135 OK 4.51080 6.43884 0.513418
22 uc001acu.3 C1orf159 NM_017891 10 1841 174 1070 OK 3.47172 8.62867 1.313490
> noncoding<-m2_filtered[grep("NR_",m2_filtered$refseq),]
> head(noncoding)
X.kgID geneSymbol refseq exon length utr5 utr3 status value_1 value_2 log2.fold_change.
137 uc001akt.5 TP73-AS1 NR_033712 6 3329 NA NA OK 0.14756 1.02529 2.796660
338 uc001axj.4 FLJ37453 NR_024279 2 2473 NA NA OK 8.08395 12.44000 0.621857
459 uc001bfn.6 LINC00339 NR_109760 2 1054 NA NA OK 7.62286 10.25620 0.428090
490 uc001bhp.3 TCEB3-AS1 NR_038280 4 1074 NA NA OK 11.70090 8.02035 -0.544877
612 uc001boy.1 SCARNA1 NR_002997 1 166 NA NA OK 26.24250 116.47800 2.150090
622 uc001bpk.4 EYA3 NR_104214 17 1806 NA NA OK 2.26155 4.16294 0.880288
> #出力用のファイルの準備
> postscript("Scatter_plot.eps", horizontal = FALSE, onefile = FALSE,paper = "special", height = 5, width = 5)
> plot(log(coding$value_1),log(coding$value_2),col=3,xlim=c(-10,10),ylim=c(-10,10),xlab="FPKM_1",ylab="FPKM_2")
> par(new=T)
> plot(log(noncoding$value_1),log(noncoding$value_2),col=4,xlim=c(-10,10),ylim=c(-10,10),xlab="FPKM_1",ylab="FPKM_2")
> #ファイルへの出力終了
> dev.off()
quartz
2
ここでcol=は色指定、xlim,ylimはx,y軸の値をベクトル指定、xlab,ylabはx,y軸のラベルの指定、par(new=T)は重ね合わせのおまじない。 これで、Scatter_plot.epsを開いてやれば、ほら、どこかで見た図が!
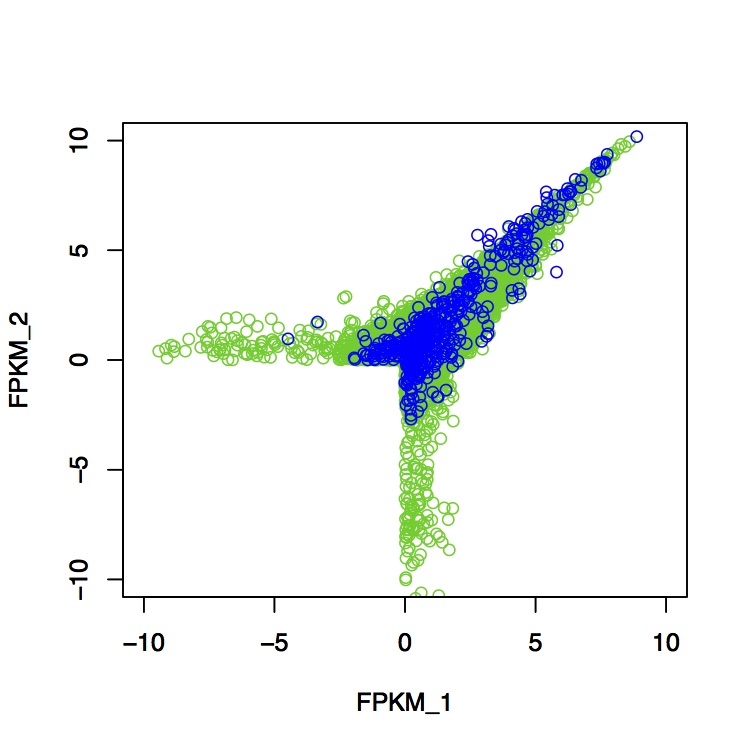
緑がcoding、青がnoncodingですね。logが負の値は発現量が低すぎるということで、cuffdiffのtest=OKのフィルターでごそっと抜けてます。転写産物の長さとfold changeの関係もついでに見てみます。
> #出力用のファイルの準備
> postscript("length.eps", horizontal = FALSE, onefile = FALSE,paper = "special", height = 5, width = 5)
> plot(coding$length,coding$log2.fold_change.,col=3,xlim=c(0,30000),ylim=c(-10,10),xlab="length",ylab="FC")
> par(new=T)
> plot(noncoding$length,noncoding$log2.fold_change.,col=4,xlim=c(0,30000),ylim=c(-10,10),xlab="length",ylab="FC")#ファイルへの出力終了
> dev.off()
quartz
2
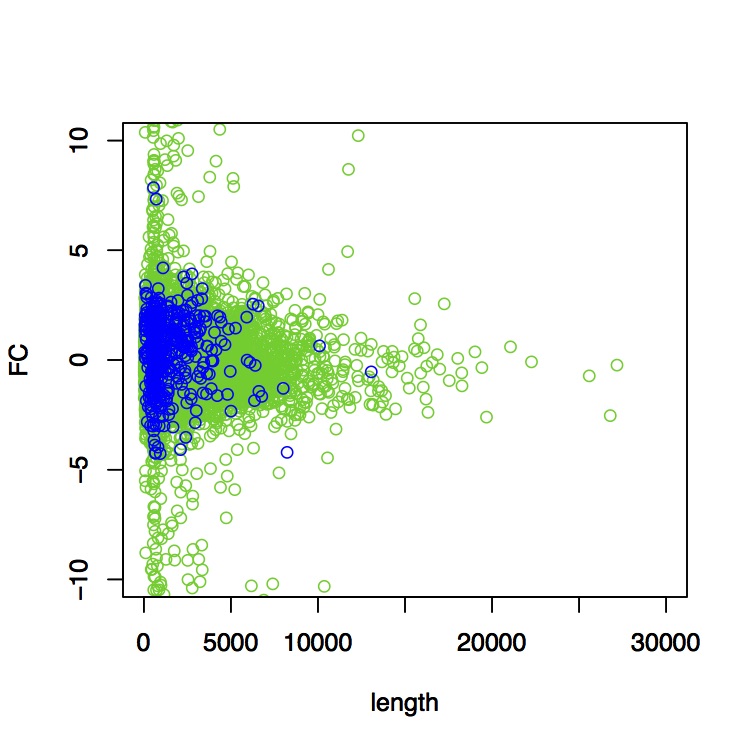
うん、なんかそれらしい図がかけた。論文にも使えそう!
以上、ピペットマンとピンセットしか握ったことのないコテコテのベンチ屋がRをちろっと使ってみた、の巻でした。プロになろう、ドライになろうと無駄にハードルを上げなくても、合言葉は「moistureで行こう!」です。GreatとShitだけ覚えとけば英語の日常会話はなんとかなるとかいう噂もありますが、RにしてもXX<-[x,y]の使い方とmergeだけ覚えといて、あとはググれば大概のことはなんとかなるような気がします。英語だって、流暢な英語で話すことよりも、まずは外に飛び出してこんな世界もあるんだ!という感動を経験することのほうがよっぽど大事です。僕自身はまだまだ幼稚園児にも届かないレベルのR語しか使えませんが、moistureな世界を垣間見て、こりゃ楽しいわい、と。バイオインフォの専門家にミニプレップレベルの仕事を丸投げするのでなく、ベンチ屋もある程度の解析は自前でこなす。そうすることで、いろいろ新しい可能性も開けてくることかと思います。特に実験系のラボにいる若い学生さんやポスドクの皆さん、何はともあれRの扉を叩いてみましょう!

 2020
2020