最近は培養細胞や組織ごとの遺伝子発現量はウェブベースの様々なサービスで簡単にわかる様になってきていますが、例えば何らかの実験操作を加えた時や、特定の変異体での発現変動を見ようと思ったら、やはり公共データベースから生データを落としてきて自前で解析、という作業は欠かせません。また、非モデル生物でもNGSデータの方はすごい勢いで充実してきてますので、そいつらを利用しない手はない。今回は、高脂肪食を与えた時の褐色脂肪での遺伝子発現ってどうなってるんだろう、と、ふと思ったので、そこから始めてみたいと思います。
まずは、悪名高きNCBIのSRAで目的のファイルを検索します。DBCLS SRAの方がダウンロードのリンクが見つけやすく使いやすくはあるのですが、最新データはやはりこちらの方が充実している様で、そちらを使ってみます。高脂肪食で褐色脂肪、ということで、検索はhigh fat mouse brownぐらいで行きましょうか。
.jpg)
いっぱい出てきましたね。同じ実験でもデータごとに別のSRAがヒットするので、上の4つは同じexperimentかな。とりあえず上から4つ目をクリックしてみます。メニューの一番上は頼みにくい。一番下も頼みにくい。この心理状態って一体なんなんでしょう?
.jpg)
心理状態はおいといて、はい。中身を見ていくと、ちょうど探していた高脂肪食時の褐色細胞での遺伝子発現変動を見たデータの様です。次に、"Study"の項目の"All runs"をクリックして、落としてきたいファイルの番号を確認します。
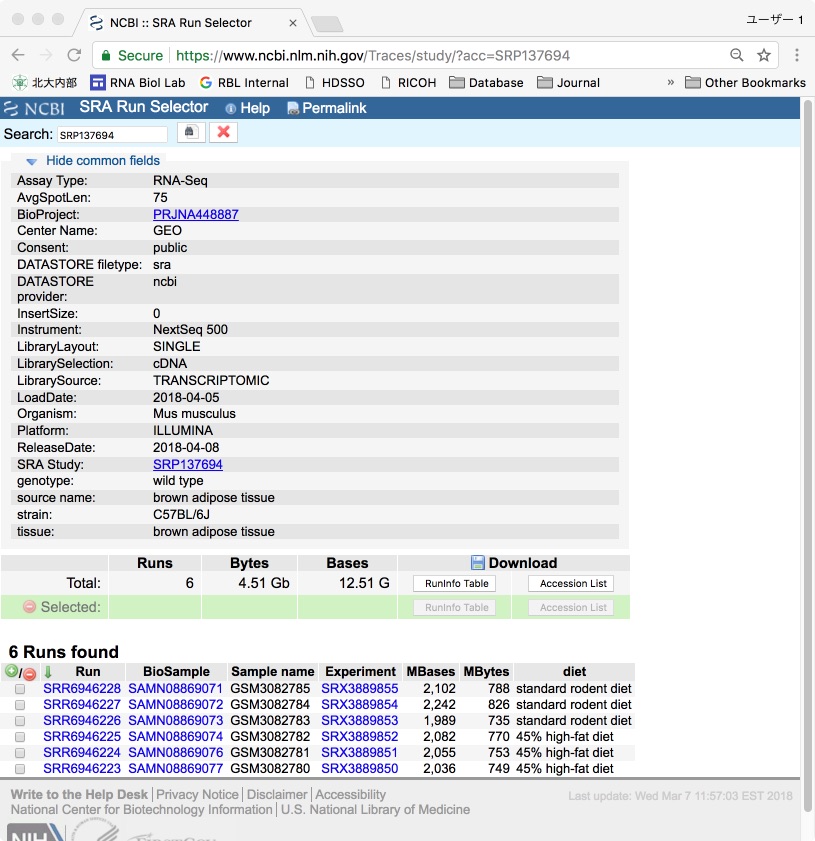
ふむふむ。SRR6946223からSRR6946228のファイルをダウンロードすれば良いわけか。さて、普通の感覚で行くとここで該当Run番号をクリックするとSRAファイルのダウンロード先のリンクが出てきても良さそうなものなのですが、このNCBIのSRAサイトが超使いにくいのは、そのリンクがものすごく見つけにくいところにあるというところで、いつものノリでクリック♪クリック♪していくと、全然目的の場所には辿り着けずにいつの間にかもとのページに戻ってきたりします。樹海か!
色々なやり方がありますが(追記:こちらのページにあるようにSRA toolsのprefetchコマンドを使うやり方はとてもシンプルでわかりやすいです。-t ascpオプションをつければAsperaもつかえるみたい。本家NCBIのページにも詳しい解説があります。)、ターミナルを使ってAspera Connectのコマンドラインで落としてくるのが、爆速かつ記録も残って良い、ということで、高速ダウンロードツール、Aspera Connectをインストールします。まずはAspera Connectのホームページに行って、ごくごく普通にアプリケーションをインストールします。Webブラウザのプラグインを使っても良いのですが、コマンドラインの使い方はAspera Connectの解説ページに書いてあって、それによれば、
/path/to/ascp -T -k 1 -i path/to/private/key anonftp@ftp.ncbi.nlm.nih.gov:/path/to/data /local/locationとあります。で、どうするんだ、、、
こんな時はマニュアル読め!マニュアル!ということで、読んでみますと、 /path/to/ascpというのは、アプリケーションの場所の指定、ということで、Aspera Connectへの絶対パスを指定。 path/to/private/keyというのはよくわからんけどkey fileの場所の指定ということで、asperaweb_id_dsa.opensshというファイルの絶対パスを指定してやるらしい。 /path/to/dataというのは落としてきたいデータのありか。これも不親切でリンク先の解説をよく見てもよくわからないのですが、本家NCBIのページに具体例がのっていて、例えばSRR6946223の場合、
/sra/sra-instant/reads/ByRun/sra/SRR/SRR694/SRR6946223/SRR6946223.sra
になります。後ろから3番目のところはファイル名の先頭から6文字を入力するようです。 /local/locationはファイルをダウンロードする場所。これを指定しないとエラーが出ます。今回はカレントディレクトリ、ということで.(ピリオド)にします。
さて、文法の解読が済んだところでさっそくコマンドをつくろうとするわけですが、ここで第二の壁。"ascp"ってどこにあるんだ?じつはこれ、Aspera Connectのアプリケーションのパッケージの中にあるのですが、普段は見えないんですよね。Macの場合、アプリケーションのアイコンを右クリックすると「パッケージの内容を表示」というオプションが出てくるので、それを選ぶと下流のフォルダ等を表示することができます。
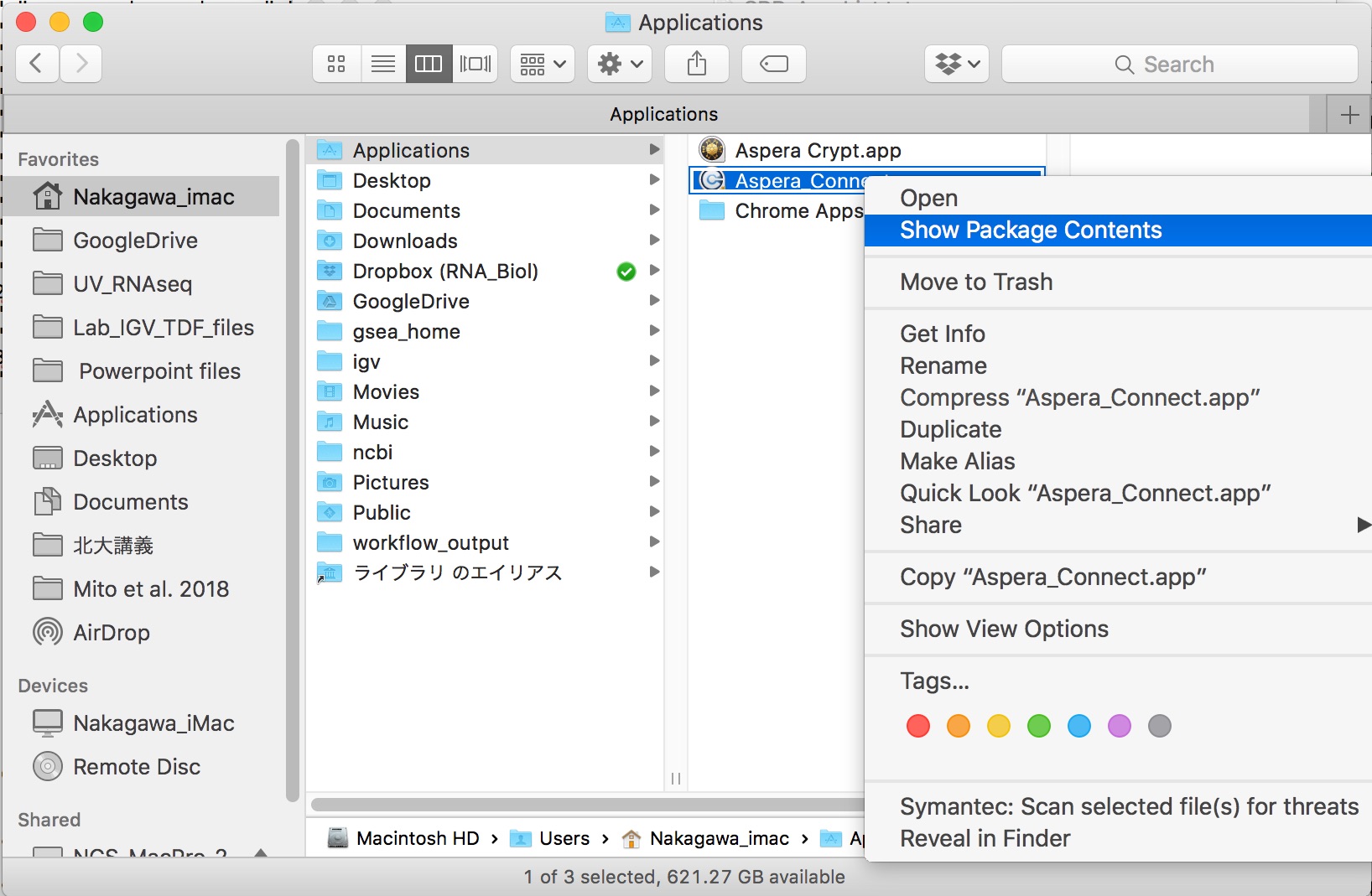
すると、、、
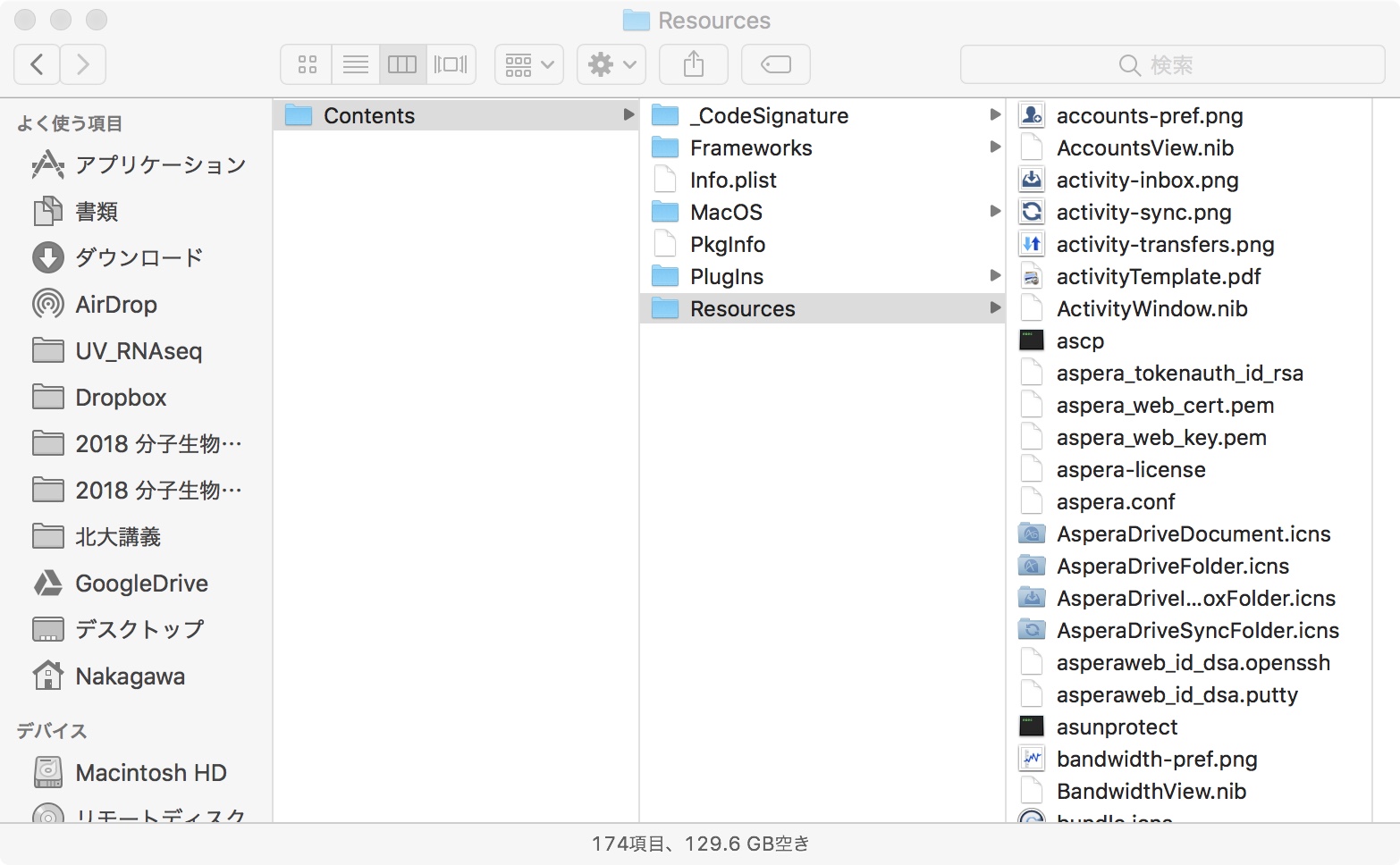
あったあった!上から8番目のやつですね。また、上から18番目に探し求めていたkey fileであるasperaweb_id_dsa.opensshも入ってます。ちなみにそもそもこのAspera Connectのアプリケーションの場所もわかりにくくて、デフォルトのままインストールすると、各自の「ホームフォルダ」の下のアプリケーションフォルダの中に入ってます。ちなみに最新のOSだと「ホームフォルダ」の場所もわかりにくくて、デフォルトのFinderの設定だと「ホームフォルダ」がサイドバーに表示されません。Finderの環境設定でサイドバーに表示する項目でホームフォルダにチェックを入れておきます。
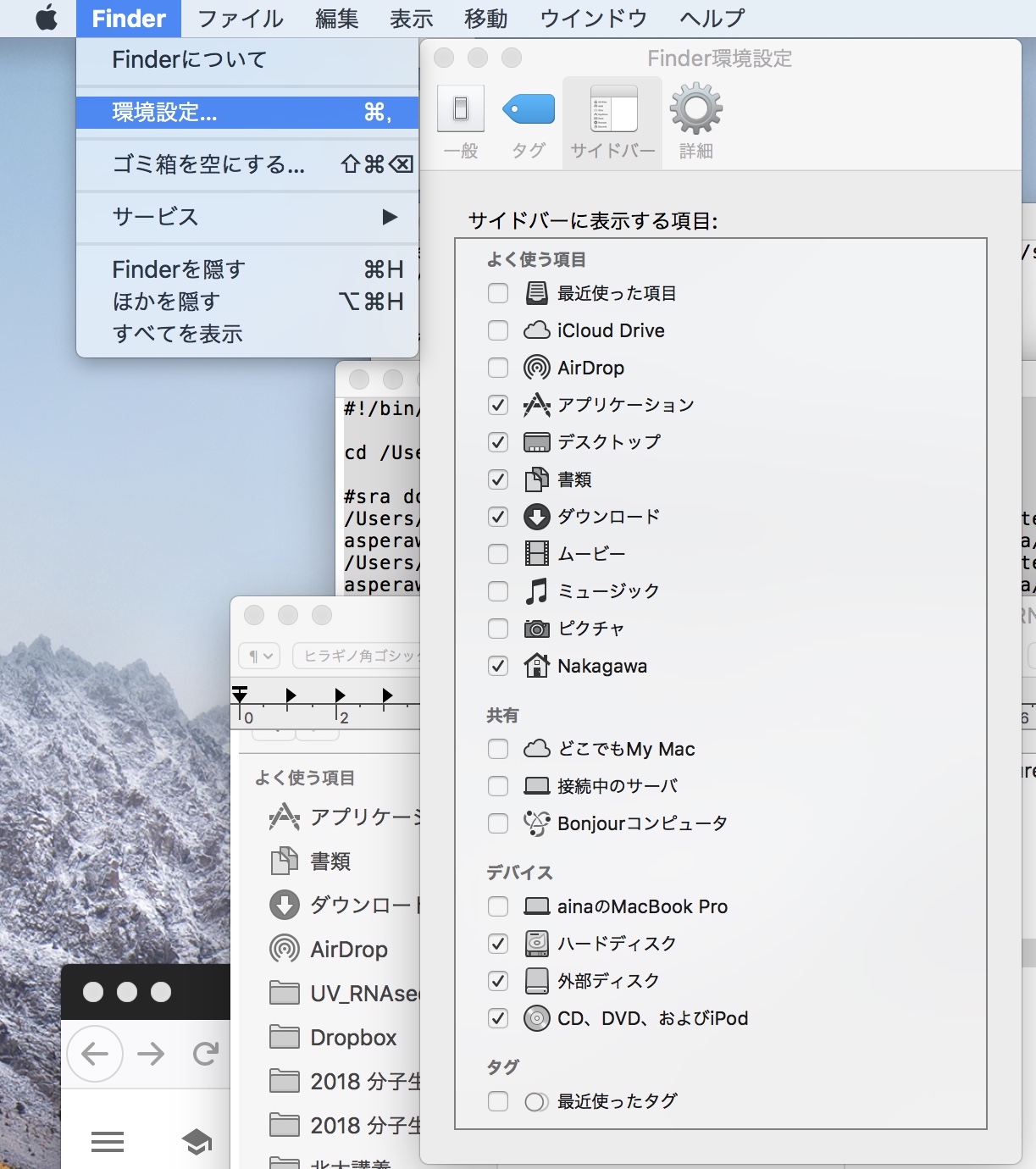
どんだけトラップの数が多いんだ。。。
気を取り直してダウンロードのためのコマンド作り。テキストエディットであらかじめコマンドを作っておいて、ターミナルに貼り付けるのが記録も残るのでGoodです。ファイルへの絶対パスの指定は手入力すると十中八九まちがえるので、「標準テキスト表示」にしたテキストエディットの新規書類にファイルのアイコンをドラッグ&ドロップで引っ張ってくるのが間違いありません。し・か・し。ここでもハニートラップ。"Aspera Connect"と、アプリケーションフォルダ名にスペースが入っているではないか!ターミナルではスペースは区切りとして認識されてしまうので、そのまえにバックスラッシュ記号\をつけなくてはなりません。たとえば今回の作業用のフォルダ2018_NGS_standardをデスクトップに作ってSRR6946223をダウンロードする場合、
cd /Users/Nakagawa_imac/Desktop/2018_NGS_standard
/Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/ascp -T -k 1 -i /Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/asperaweb_id_dsa.openssh anonftp@ftp.ncbi.nlm.nih.gov:/sra/sra-instant/reads/ByRun/sra/SRR/SRR694/SRR6946223/SRR6946223.sra .になります。最後のピリオド、保存先の指定なので忘れてはいけません。ダメ、絶対です。なんだか数々の地雷をさけながらすごく遠い道のりをはるばるきた感じがしますが、一度この文さえ作ってしまえば、あとは"SRR6946223"の部分を検索・自分が落としたいIDで置換すれば良いだけ。意外と手間はかかりません。パス指定の最後から3番目のSRR694の部分は検索ではひっかかってこないので、そこだけ注意です。
さてと。この爆速Aspera Connect、本当に早いです。ftpサイトから普通に落としてくるのに比べたら、路面電車と新幹線ぐらいの差はあります。なんでこれまで使っていなかったんだろうと、後悔しきり。。。
お次はマッピングです。sraはマッピングツールが食べてくれないので、fastqに変換します。おなじみsratoolkitですね。sratoolkitをhomebrewでインストールしておき(追記: brew install sratoolkitとやろうとしてXcode command line toolがないと怒られたときは。Xcodeのインストールをしてからになります)、今回はシングルエンドなのでシンプルに
fastq-dump --gzip ./SRR6946223.sraでOK。下記の遠藤さんのコメントにありますが、--gzipオプションをつけとくと割とでかいfastqファイルを圧縮したgzipで扱えるので便利なようです。ありがたやありがたや。このfastq.gzファイルを、HISAT2に食わせます。HISAT2の使い方はmacでインフォマティクスさんのページが詳しいです。初めてHISAT2を使う時は、検索用のインデックスファイルを作らなくてはいけません。せっかくですからゲノムファイルも最新のを、ということで、2018_NGS_standardフォルダの下にgenomeフォルダを作って、GENCODEの最新マウスゲノムGenome sequence (GRCm38.p6)をダウンロードしておきます。ファイル名はGRCm38.p6.genome.faですね。そしておなじみのhomebrewでhisat2をインストール。
brew install hisat2追記: homebrewのインストールで若干引っかかる可能性があるみたいです。rnacintosh LC475さんご指摘ありがとうございます。
そうなんですよ。Homebrew/scienceがbrewsci/bioに移動されたためhisat2が行方不明になっていたという問題でした。
— rnacintosh LC475 (@68040LC) June 6, 2018
brew tap brewsci/bio
brew install hisat2
で解決します。
参考https://t.co/J5DNYZVtiI
次に、インデックスを作成。genome_indexという名前を共通でつけときます。
hisat2-build GRCm38.p6.genome.fa genome_indexここが一番時間がかかるステップですが、一度やっておけば、当然ながらもうやる必要はありません。準備が整ったところでHISAT2にfastqファイルとゲノムのインデックスを投げてやります。HISAT2はtophat2の進化形。こちらは路面電車が飛行機に進化したぐらいの差があるようです。まさに爆速。一晩かかっていたマッピングが数分で終わります。今までの時間はなんだったんだ、、、ここでは僕のiMacに合わせてコア数を4に指定しています。今回のfastqはシングルエンドなので、-Uでfastqを指定してます。
hisat2 -p 4 -x ./genome/genome_index -U SRR6946223.fastq.gz -S SRR6946223.samあとはこれをソートして、bamに変えて、インデックスをつければ完成!二階堂さんのページを参考に、粛々と。
samtools sort -@ 4 -O bam -o SRR6946223.sort.bam SRR6946223.sam
samtools index SRR6946223.sort.bam一度環境を作ってしまえば、数ギガのfastqでも10分そこらでマッピングまで完了してしまうので、シェルスクリプトを作ってちょこちょこやれば、きつねどん兵衛を電子レンジでチンして食べ終わる頃にはIGVで見れる、というわけです。まさにAsperaConnectとHISAT2さまさま、です。次回はcuffdiffでなくfeatureCountsとedgeRをつかったなんちゃってバイオインフォ解析に移りたいと思います。今回のマッピングの一連の作業を6ファイルすべてについておこなうスクリプトを貼っときます。下の方のdownload_mapping.sh.txtのリンクからでもダウンロードできます。作業フォルダはデスクトップに作った2018_NGS_standardフォルダ。hisatのインデックスはその下に作ったgenomeファイルにgenome_indexの名前で作ったのを入れてます。
追記:下記のawkマジシャン遠藤さんのコメントで、ループを使えば簡単!ということでしたので、そちらもやってみました。二つのスクリプトはやってることは同じです。ありがとうございました!
#!/bin/sh
cd /Users/Nakagawa_imac/Desktop/2018_NGS_standard
#set loop
for srr in SRR69462{23..28}
do
#sra download
/Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/ascp -T -k 1 -i /Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/asperaweb_id_dsa.openssh anonftp@ftp.ncbi.nlm.nih.gov:/sra/sra-instant/reads/ByRun/sra/SRR/${srr::6}/${srr}/${srr}.sra .
#sam to bam conversinon
fastq-dump --gzip ./${srr}.sra
#hisat2 mapping
hisat2 -p 4 -x ./genome/genome_index -U ${srr}.fastq.gz -S ${srr}.sam
#sam to sorted bam
samtools sort -@ 4 -O bam -o ${srr}.sort.bam ${srr}.sam
#create index
samtools index ${srr}.sort.bam
done
#!/bin/sh
cd /Users/Nakagawa_imac/Desktop/2018_NGS_standard
#sra download
/Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/ascp -T -k 1 -i /Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/asperaweb_id_dsa.openssh anonftp@ftp.ncbi.nlm.nih.gov:/sra/sra-instant/reads/ByRun/sra/SRR/SRR694/SRR6946223/SRR6946223.sra .
/Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/ascp -T -k 1 -i /Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/asperaweb_id_dsa.openssh anonftp@ftp.ncbi.nlm.nih.gov:/sra/sra-instant/reads/ByRun/sra/SRR/SRR694/SRR6946224/SRR6946224.sra .
/Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/ascp -T -k 1 -i /Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/asperaweb_id_dsa.openssh anonftp@ftp.ncbi.nlm.nih.gov:/sra/sra-instant/reads/ByRun/sra/SRR/SRR694/SRR6946225/SRR6946225.sra .
/Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/ascp -T -k 1 -i /Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/asperaweb_id_dsa.openssh anonftp@ftp.ncbi.nlm.nih.gov:/sra/sra-instant/reads/ByRun/sra/SRR/SRR694/SRR6946226/SRR6946226.sra .
/Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/ascp -T -k 1 -i /Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/asperaweb_id_dsa.openssh anonftp@ftp.ncbi.nlm.nih.gov:/sra/sra-instant/reads/ByRun/sra/SRR/SRR694/SRR6946227/SRR6946227.sra .
/Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/ascp -T -k 1 -i /Users/Nakagawa_imac/Applications/Aspera\ Connect.app/Contents/Resources/asperaweb_id_dsa.openssh anonftp@ftp.ncbi.nlm.nih.gov:/sra/sra-instant/reads/ByRun/sra/SRR/SRR694/SRR6946228/SRR6946228.sra .
#sam to bam conversinon
fastq-dump ./SRR6946223.sra
fastq-dump ./SRR6946224.sra
fastq-dump ./SRR6946225.sra
fastq-dump ./SRR6946226.sra
fastq-dump ./SRR6946227.sra
fastq-dump ./SRR6946228.sra
#hisat2 mapping
hisat2 -p 4 -x ./genome/genome_index -U SRR6946223.fastq -S SRR6946223.sam
hisat2 -p 4 -x ./genome/genome_index -U SRR6946224.fastq -S SRR6946224.sam
hisat2 -p 4 -x ./genome/genome_index -U SRR6946225.fastq -S SRR6946225.sam
hisat2 -p 4 -x ./genome/genome_index -U SRR6946226.fastq -S SRR6946226.sam
hisat2 -p 4 -x ./genome/genome_index -U SRR6946227.fastq -S SRR6946227.sam
hisat2 -p 4 -x ./genome/genome_index -U SRR6946228.fastq -S SRR6946228.sam
#sam to sorted bam & index
samtools sort -@ 4 -O bam -o SRR6946223.sort.bam SRR6946223.sam
samtools sort -@ 4 -O bam -o SRR6946224.sort.bam SRR6946224.sam
samtools sort -@ 4 -O bam -o SRR6946225.sort.bam SRR6946225.sam
samtools sort -@ 4 -O bam -o SRR6946226.sort.bam SRR6946226.sam
samtools sort -@ 4 -O bam -o SRR6946227.sort.bam SRR6946227.sam
samtools sort -@ 4 -O bam -o SRR6946228.sort.bam SRR6946228.sam
samtools index SRR6946223.sort.bam
samtools index SRR6946224.sort.bam
samtools index SRR6946225.sort.bam
samtools index SRR6946226.sort.bam
samtools index SRR6946227.sort.bam
samtools index SRR6946228.sort.bam

 2020
2020