
Tropomyosin, one of major actin-filament binding proteins, regulates actin-myosin interaction and actin filament stability. Multicellular organisms express a number of tropomyosin isoforms, but understanding of isoform-specific tropomyosin functions is incomplete. The nematode Caenorhabditis elegans has a single tropomyosin gene, lev-11, which has been reported to express four isoforms by using two separate promoters and alternative splicing. Here, we report a fifth tropomyosin isoform, LEV-11O, which is produced by alternative splicing which includes a newly identified seventh exon, exon 7a. By visualizing specific splicing events in vivo, we find that exon 7a is predominantly selected in a subset of the body wall muscles in the head, while exon 7b, which is alternative to exon 7a, is utilized in the rest of the body. Point mutations in exon 7a and exon 7b cause resistance to levamisole-induced muscle contraction specifically in the head and the main body, respectively. Overexpression of LEV-11O, but not LEV-11A, in the main body results in weak levamisole resistance. These results demonstrate that specific tropomyosin isoforms are expressed in the head and body regions of the muscles and differentially contribute to the regulation of muscle contractility.
The nervous system of higher eukaryotes is composed of numerous types of neurons and glia that together orchestrate complex neuronal responses. However, this complex pool of cells typically poses analytical challenges in investigating gene expression profiles and their epigenetic basis for specific cell types. Here, we developed a novel method that enables cell type-specific analyses of epigenetic modifications using tandem chromatin immunoprecipitation sequencing (tChIP-Seq). FLAG-tagged histone H2B, a constitutive chromatin component, was first expressed in Camk2a-positive pyramidal cortical neurons and used to purify chromatin in a cell type-specific manner. Subsequent chromatin immunoprecipitation using antibodies against H3K4me3-a chromatin modification mainly associated with active promoters-allowed us to survey the histone modifications in Camk2a-positive neurons. Indeed, tChIP-Seq identified hundreds of H3K4me3 modifications in promoter regions located upstream of genes associated with neuronal functions and genes with unknown functions in cortical neurons. tChIP-Seq provides a versatile approach to investigating the epigenetic modifications of particular cell types in vivo.
Long noncoding RNA (lncRNA) molecules are some of the newest and least understood players in gene regulation. Hence, we need good model systems with well-defined RNA and protein components. One such system is paraspeckles - protein-rich nuclear organelles built around a specific lncRNA scaffold. New discoveries show how paraspeckles are formed through multiple RNA-protein and protein-protein interactions, some of which involve extensive polymerization, and others with multivalent interactions driving phase separation. Once formed, paraspeckles influence gene regulation through sequestration of component proteins and RNAs, with subsequent depletion in other compartments. Here we focus on the dual aspects of paraspeckle structure and function, revealing an emerging role for these dynamic bodies in a multitude of cellular settings.
The epigenetic phenomenon called X chromosome inactivation plays critical roles in female development in eutherian mammals, and has attracted attention in the fields of developmental biology and regenerative biology in efforts to understand the pluripotency of stem cells. X chromosome inactivation is routinely studied after cell fixation, but live imaging is increasingly being required to improve our understanding of the dynamics and kinetics of X chromosome inactivation and reactivation processes. Here, we describe our live imaging method to monitor the epigenetic status of X chromosomes using a gene knock-in mouse strain named "Momiji" and give an overview of the application of this strain as a resource for biological and stem cell research.
It has been 8 years since the concept of naïve and primed pluripotent stem cell states was first proposed. Both are states of pluripotency, but exhibit slightly different properties. The naïve state represents the cellular state of the preimplantation mouse blastocyst inner cell mass, while the primed state is representative of the post-implantation epiblast cells. These two cell types exhibit clearly distinct developmental potential, as evidenced by the fact that naïve cells are able to contribute to blastocyst chimeras, while primed cells cannot. However, the epigenetic differences that underlie the distinct developmental potential of these cell types remain unclear, which is rather surprising given the large amount of active investigation over the years. Elucidating such epigenetic differences should lead to a better understanding of the fundamental properties of these states of pluripotency and the means by which the naïve-to-primed transition occurs, which may provide insights into the essence of stem cell commitment.
The CRISPR-associated endonuclease Cas9 binds to a guide RNA and cleaves doublestranded DNA with a sequence complementary to the RNA guide. The Cas9–RNA system has been harnessed for numerous applications, such as genome editing. Here we use high-speed atomic force microscopy (HS-AFM) to visualize the real-space and real-time dynamics of CRISPR-Cas9 in action. HS-AFM movies indicate that, whereas apo-Cas9 adopts unexpected flexible conformations, Cas9–RNA forms a stable bilobed structure and interrogates target sites on the DNA by three-dimensional diffusion. These movies also provide real-time visualization of the Cas9-mediated DNA cleavage process. Notably, the Cas9 HNH nuclease domain fluctuates upon DNA binding, and subsequently adopts an active conformation, where the HNH active site is docked at the cleavage site in the target DNA. Collectively, our HS-AFM data extend our understanding of the action mechanism of CRISPR-Cas9.
X inactive-specific transcript (Xist) is a long noncoding RNA that plays an essential role in X chromosome inactivation. Although Xist RNA, like common protein-coding mRNAs, is transcribed by RNA polymerase II, spliced and polyadenylated, it is retained in the nucleus and associates with the X chromosome it originates from. It has been assumed that Xist RNA recruits proteins involved in epigenetic modifications and chromatin compaction to the X chromosome. One of the major proteins constituting the nuclear matrix, hnRNP U, has been shown to be required for the association of Xist RNA with the inactive X chromosome (Xi). In this study, we found that the first 950-nt sequence of Xist RNA had the potential to associate with chromatin in a manner independent of hnRNP U. Furthermore, its chromatin association is apparently dependent on the presence of an intact A-repeat sequence, which is one of the repeats in Xist/XIST RNA conserved among many mammalian species, and has been shown to be important for Xist RNA-mediated silencing. Taking this unexpected finding and a previous study demonstrating the effect of Xist RNA lacking the A-repeat on the formation of the silent heterochromatin domain together, we suggest that the A-repeat captures chromatin near the initial loading site of Xist RNA and relocates it into the core of the heterochromatin domain.
Xist RNA, which is responsible for X inactivation, is a key epigenetic player in the embryogenesis of female mammals. Of the several repeats conserved in Xist RNA, the A-repeat has been shown to be essential for its silencing function in differentiating embryonic stem cells. Here, we introduced a new Xist allele into mouse that produces mutated Xist RNA lacking the A-repeat (XistCAGΔ5' ). XistCAGΔ5' RNA expressed in the embryo coated the X chromosome but failed to silence it. Although imprinted X inactivation was substantially compromised upon paternal transmission, allele-specific RNA-seq in the trophoblast revealed that XistCAGΔ5' RNA still retained some silencing ability. Furthermore, the failure of imprinted X inactivation had more significant impacts than expected on genome-wide gene expression. It is likely that dosage compensation is required not only for equalizing X-linked gene expression between the sexes but also for proper global gene regulation in differentiated female somatic cells.
Cell type-specific transcriptomes are enabled by the action of multiple regulators, which are frequently expressed within restricted tissue regions. In the present study, we identify one such regulator, Quaking 5 (Qki5), as an RNA-binding protein (RNABP) that is expressed in early embryonic neural stem cells and subsequently down-regulated during neurogenesis. mRNA sequencing analysis in neural stem cell culture indicates that Qki proteins play supporting roles in the neural stem cell transcriptome and various forms of mRNA processing that may result from regionally restricted expression and subcellular localization. Also, our in utero electroporation gain-of-function study suggests that the nuclear-type Qki isoform Qki5 supports the neural stem cell state. We next performed in vivo transcriptome-wide protein-RNA interaction mapping to search for direct targets of Qki5 and elucidate how Qki5 regulates neural stem cell function. Combined with our transcriptome analysis, this mapping analysis yielded a bona fide map of Qki5-RNA interaction at single-nucleotide resolution, the identification of 892 Qki5 direct target genes, and an accurate Qki5-dependent alternative splicing rule in the developing brain. Last, our target gene list provides the first compelling evidence that Qki5 is associated with specific biological events; namely, cell-cell adhesion. This prediction was confirmed by histological analysis of mice in which Qki proteins were genetically ablated, which revealed disruption of the apical surface of the lateral wall in the developing brain. These data collectively indicate that Qki5 regulates communication between neural stem cells by mediating numerous RNA processing events and suggest new links between splicing regulation and neural stem cell states.
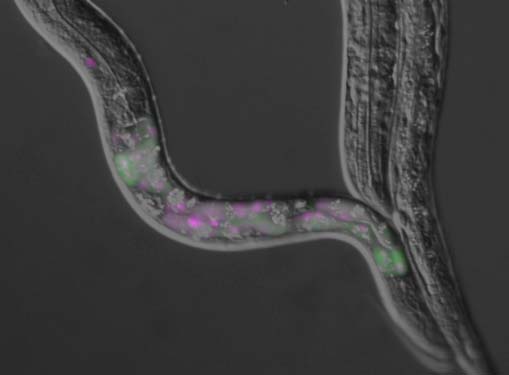 A nematode Caenorhabditis elegans is an intron-rich organism and up to 25% of its pre-mRNAs are estimated to be alternatively processed. Its compact genomic organization enables construction of fluorescence splicing reporters with intact genomic sequences and visualization of alternative processing patterns of interest in the transparent living animals with single-cell resolution. Genetic analysis with the reporter worms facilitated identification of trans-acting factors and cis-acting elements, which are highly conserved in mammals. Analysis of unspliced and partially spliced pre-mRNAs in vivo raised models for alternative splicing regulation relying on specific order of intron excision. RNA-seq analysis of splicing factor mutants and CLIP-seq analysis of the factors allow global search for target genes in the whole animal. An mRNA surveillance system is not essential for its viability or fertility, allowing analysis of unproductively spliced noncoding mRNAs. These features offer C. elegans as an ideal model organism for elucidating alternative pre-mRNA processing mechanisms in vivo. Examples of isoform-specific functions of alternatively processed genes are summarized.
A nematode Caenorhabditis elegans is an intron-rich organism and up to 25% of its pre-mRNAs are estimated to be alternatively processed. Its compact genomic organization enables construction of fluorescence splicing reporters with intact genomic sequences and visualization of alternative processing patterns of interest in the transparent living animals with single-cell resolution. Genetic analysis with the reporter worms facilitated identification of trans-acting factors and cis-acting elements, which are highly conserved in mammals. Analysis of unspliced and partially spliced pre-mRNAs in vivo raised models for alternative splicing regulation relying on specific order of intron excision. RNA-seq analysis of splicing factor mutants and CLIP-seq analysis of the factors allow global search for target genes in the whole animal. An mRNA surveillance system is not essential for its viability or fertility, allowing analysis of unproductively spliced noncoding mRNAs. These features offer C. elegans as an ideal model organism for elucidating alternative pre-mRNA processing mechanisms in vivo. Examples of isoform-specific functions of alternatively processed genes are summarized.