-
Home

- Publication

カテゴリ一覧から年度または研究者名を選ぶと絞り込みができます。
Choose the fiscal year (FY) or the researcher name from the category list.
Xist RNA, which is responsible for X inactivation, is a key epigenetic player in the embryogenesis of female mammals. Of the several repeats conserved in Xist RNA, the A-repeat has been shown to be essential for its silencing function in differentiating embryonic stem cells. Here, we introduced a new Xist allele into mouse that produces mutated Xist RNA lacking the A-repeat (XistCAGΔ5' ). XistCAGΔ5' RNA expressed in the embryo coated the X chromosome but failed to silence it. Although imprinted X inactivation was substantially compromised upon paternal transmission, allele-specific RNA-seq in the trophoblast revealed that XistCAGΔ5' RNA still retained some silencing ability. Furthermore, the failure of imprinted X inactivation had more significant impacts than expected on genome-wide gene expression. It is likely that dosage compensation is required not only for equalizing X-linked gene expression between the sexes but also for proper global gene regulation in differentiated female somatic cells.
Cell type-specific transcriptomes are enabled by the action of multiple regulators, which are frequently expressed within restricted tissue regions. In the present study, we identify one such regulator, Quaking 5 (Qki5), as an RNA-binding protein (RNABP) that is expressed in early embryonic neural stem cells and subsequently down-regulated during neurogenesis. mRNA sequencing analysis in neural stem cell culture indicates that Qki proteins play supporting roles in the neural stem cell transcriptome and various forms of mRNA processing that may result from regionally restricted expression and subcellular localization. Also, our in utero electroporation gain-of-function study suggests that the nuclear-type Qki isoform Qki5 supports the neural stem cell state. We next performed in vivo transcriptome-wide protein-RNA interaction mapping to search for direct targets of Qki5 and elucidate how Qki5 regulates neural stem cell function. Combined with our transcriptome analysis, this mapping analysis yielded a bona fide map of Qki5-RNA interaction at single-nucleotide resolution, the identification of 892 Qki5 direct target genes, and an accurate Qki5-dependent alternative splicing rule in the developing brain. Last, our target gene list provides the first compelling evidence that Qki5 is associated with specific biological events; namely, cell-cell adhesion. This prediction was confirmed by histological analysis of mice in which Qki proteins were genetically ablated, which revealed disruption of the apical surface of the lateral wall in the developing brain. These data collectively indicate that Qki5 regulates communication between neural stem cells by mediating numerous RNA processing events and suggest new links between splicing regulation and neural stem cell states.
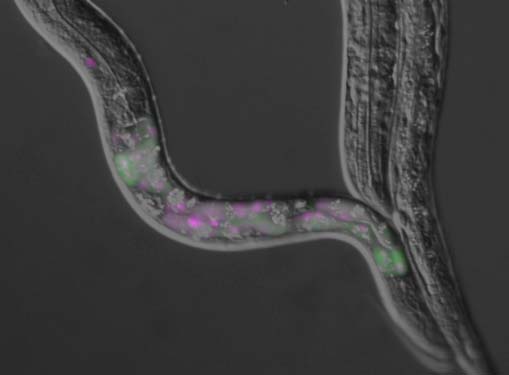 A nematode Caenorhabditis elegans is an intron-rich organism and up to 25% of its pre-mRNAs are estimated to be alternatively processed. Its compact genomic organization enables construction of fluorescence splicing reporters with intact genomic sequences and visualization of alternative processing patterns of interest in the transparent living animals with single-cell resolution. Genetic analysis with the reporter worms facilitated identification of trans-acting factors and cis-acting elements, which are highly conserved in mammals. Analysis of unspliced and partially spliced pre-mRNAs in vivo raised models for alternative splicing regulation relying on specific order of intron excision. RNA-seq analysis of splicing factor mutants and CLIP-seq analysis of the factors allow global search for target genes in the whole animal. An mRNA surveillance system is not essential for its viability or fertility, allowing analysis of unproductively spliced noncoding mRNAs. These features offer C. elegans as an ideal model organism for elucidating alternative pre-mRNA processing mechanisms in vivo. Examples of isoform-specific functions of alternatively processed genes are summarized.
A nematode Caenorhabditis elegans is an intron-rich organism and up to 25% of its pre-mRNAs are estimated to be alternatively processed. Its compact genomic organization enables construction of fluorescence splicing reporters with intact genomic sequences and visualization of alternative processing patterns of interest in the transparent living animals with single-cell resolution. Genetic analysis with the reporter worms facilitated identification of trans-acting factors and cis-acting elements, which are highly conserved in mammals. Analysis of unspliced and partially spliced pre-mRNAs in vivo raised models for alternative splicing regulation relying on specific order of intron excision. RNA-seq analysis of splicing factor mutants and CLIP-seq analysis of the factors allow global search for target genes in the whole animal. An mRNA surveillance system is not essential for its viability or fertility, allowing analysis of unproductively spliced noncoding mRNAs. These features offer C. elegans as an ideal model organism for elucidating alternative pre-mRNA processing mechanisms in vivo. Examples of isoform-specific functions of alternatively processed genes are summarized.
The RNA-guided Cpf1 (also known as Cas12a) nuclease associates with a CRISPR RNA (crRNA) and cleaves the double-stranded DNA target complementary to the crRNA guide. The two Cpf1 orthologs from Acidaminococcus sp. (AsCpf1) and Lachnospiraceae bacterium (LbCpf1) have been harnessed for eukaryotic genome editing. Cpf1 requires a specific nucleotide sequence, called a protospacer adjacent motif (PAM), for target recognition. Besides the canonical TTTV PAM, Cpf1 recognizes suboptimal C-containing PAMs. Here, we report four crystal structures of LbCpf1 in complex with the crRNA and its target DNA containing either TTTA, TCTA, TCCA, or CCCA as the PAM. These structures revealed that, depending on the PAM sequences, LbCpf1 undergoes conformational changes to form altered interactions with the PAM-containing DNA duplexes, thereby achieving the relaxed PAM recognition. Collectively, the present structures advance our mechanistic understanding of the PAM-dependent, crRNA-guided DNA cleavage by the Cpf1 family nucleases.
The terminal uridylyltransferase, TUT1, builds or repairs the 3′-oligo-uridylylated tail of U6 snRNA. The 3′-oligo-uridylylated tail is the Lsm-binding site for U4/U6 di-snRNP formation and U6 snRNA recycling for pre-mRNA splicing. Here, we report crystallographic and biochemical analyses of human TUT1, which revealed the mechanisms for the specific uridylylation of the 3′-end of U6 snRNA by TUT1. The O2 and O4 atoms of the UTP base form hydrogen bonds with the conserved His and Asn in the catalytic pocket, respectively, and TUT1 preferentially incorporates UMP onto the 3′-end of RNAs. TUT1 recognizes the entire U6 snRNA molecule by its catalytic domains, N-terminal RNA-recognition motifs and a previously unidentified C-terminal RNA-binding domain. Each domain recognizes specific regions within U6 snRNA, and the recognition is coupled with the domain movements and U6 snRNA structural changes. Hence, TUT1 functions as the U6 snRNA-specific terminal uridylyltransferase required for pre-mRNA splicing.
Human RNA methyltransferase BCDIN3D is overexpressed in breast cancer cells, and is related to the tumorigenic phenotype and poor prognosis of breast cancer. Here, we show that cytoplasmic tRNAHis is the primary target of BCDIN3D in human cells. Recombinant human BCDIN3D, expressed in Escherichia coli, monomethylates the 5΄-monophosphate of cytoplasmic tRNAHis efficiently in vitro. In BCDN3D-knockout cells, established by CRISPR/Cas9 editing, the methyl moiety at the 5΄-monophosphate of cytoplasmic tRNAHis is lost, and the exogenous expression of BCDIN3D in the knockout cells restores the modification in cytoplasmic tRNAHis. BCIDN3D recognizes the 5΄-guanosine nucleoside at position -1 (G-1) and the eight-nucleotide acceptor helix with the G-1-A73 mis-pair at the top of the acceptor stem of cytoplasmic tRNAHis, which are exceptional structural features among cytoplasmic tRNA species. While the monomethylation of the 5΄-monophosphate of cytoplasmic tRNAHis affects neither the overall aminoacylation process in vitro nor the steady-state level of cytoplasmic tRNAHisin vivo, it protects the cytoplasmic tRNAHis transcript from degradation in vitro. Thus, BCDIN3D acts as a cytoplasmic tRNAHis-specific 5΄-methylphosphate capping enzyme. The present results also suggest the possible involvement of the monomethylation of the 5΄-monophosphate of cytoplasmic tRNAHis and/or cytoplasmic tRNAHis itself in the tumorigenesis of breast cancer cells.
The RNA-guided endonuclease Cpf1 is a promising tool for genome editing in eukaryotic cells. However, the utility of the commonly used Acidaminococcus sp. BV3L6 Cpf1 (AsCpf1) and Lachnospiraceae bacterium ND2006 Cpf1 (LbCpf1) is limited by their requirement of a TTTV protospacer adjacent motif (PAM) in the DNA substrate. To address this limitation, we performed a structure-guided mutagenesis screen to increase the targeting range of Cpf1. We engineered two AsCpf1 variants carrying the mutations S542R/K607R and S542R/K548V/ N552R, which recognize TYCV and TATV PAMs, respectively, with enhanced activities in vitro and in human cells. Genomewide assessment of off-target activity using BLISS indicated that these variants retain high DNA-targeting specificity, which we further improved by introducing an additional non-PAM-interacting mutation. Introducing the identified PAM-interacting mutations at their corresponding positions in LbCpf1 similarly altered its PAM specificity. Together, these variants increase the targeting range of Cpf1 by approximately threefold in human coding sequences to one cleavage site per ~11 bp.
The RNA-guided Cpf1 nuclease cleaves double-stranded DNA targets complementary to the CRISPR RNA (crRNA), and it has been harnessed for genome editing technologies. Recently, Acidaminococcus sp. BV3L6 (AsCpf1) was engineered to recognize altered DNA sequences as the protospacer adjacent motif (PAM), thereby expanding the target range of Cpf1-mediated genome editing. Whereas wild-type AsCpf1 recognizes the TTTV PAM, the RVR (S542R/K548V/N552R) and RR (S542R/K607R) variants can efficiently recognize the TATV and TYCV PAMs, respectively. However, their PAM recognition mechanisms remained unknown. Here we present the 2.0 Å resolution crystal structures of the RVR and RR variants bound to a crRNA and its target DNA. The structures revealed that the RVR and RR variants primarily recognize the PAM-complementary nucleotides via the substituted residues. Our high-resolution structures delineated the altered PAM recognition mechanisms of the AsCpf1 variants, providing a basis for the further engineering of CRISPR-Cpf1.
NEAT1_2 long noncoding RNA (lncRNA) is the molecular scaffold of paraspeckle nuclear bodies. Here, we report an improved RNA extraction method: extensive needle shearing or heating of cell lysate in RNA extraction reagent improved NEAT1_2 extraction by 20‐fold (a property we term “semi‐extractability”), whereas using a conventional method NEAT1_2 was trapped in the protein phase. The improved extraction method enabled us to estimate that approximately 50 NEAT1_2 molecules are present in a single paraspeckle. Another architectural lncRNA, IGS16, also exhibited similar semi‐extractability. A comparison of RNA‐seq data from needle‐sheared and control samples revealed the existence of multiple semi‐extractable RNAs, many of which were localized in subnuclear granule‐like structures. The semi‐extractability of NEAT1_2 correlated with its association with paraspeckle proteins and required the prion‐like domain of the RNA‐binding protein FUS. This observation suggests that tenacious RNA–protein and protein–protein interactions, which drive nuclear body formation, are responsible for semi‐extractability. Our findings provide a foundation for the discovery of the architectural RNAs that constitute nuclear bodies.
miRNAs play critical roles in various biological processes by targeting specific mRNAs. Current approaches to identifying miRNA targets are insufficient for elucidation of a miRNA regulatory network. Here, we created a cell-based screening system using a luciferase reporter library composed of 4,891 full-length cDNAs, each of which was integrated into the 3' UTR of a luciferase gene. Using this reporter library system, we conducted a screening for targets of miR-34a, a tumor-suppressor miRNA. We identified both previously characterized and previously uncharacterized targets. miR-34a overexpression in MDA-MB-231 breast cancer cells repressed the expression of these previously unrecognized targets. Among these targets, GFRA3 is crucial for MDA-MB-231 cell growth, and its expression correlated with the overall survival of patients with breast cancer. Furthermore, GFRA3 was found to be directly regulated by miR-34a via its coding region. These data show that this system is useful for elucidating miRNA functions and networks.